阿米卡星生產過程中雜質E形成條件探討及雜質控制策略
2021-07-12 09:01:56舒靖能廖玉華盛衛星浙江金華康恩貝生物制藥有限公司浙江金華321016
化工管理 2021年17期
關鍵詞:方法
舒靖能,廖玉華,盛衛星(浙江金華康恩貝生物制藥有限公司 ,浙江 金華 321016)
0 引言
阿米卡星,化學名稱為O-3-氨基-3-脫氧-a-D-葡吡喃糖基-(1→6)-O-[6-氨基-6-脫氧-a-D-葡吡喃糖基-(1→4)]-N-(4-氨基-2-羥基-1-氧丁基)-2-脫氧-D-鏈霉胺,為半合成氨基糖甙類抗生素,以卡那霉素為起始物料合成而來。其作用機制是作用于細菌體內的核糖體,抑制細菌蛋白質合成,并破壞細菌細胞壁的完整性,致使細菌細胞膜破壞,細胞死亡,主要依靠于細菌30S亞基結合,阻斷細菌蛋白質合成而起到抗菌作用,適用于革蘭陰性桿菌和對青霉素耐藥的金黃色葡萄球菌引起的感染。目前臨床上應用比較廣泛,有著很好的市場前景。隨著一致性評價持續進行,客戶的需求不斷提升,而且中國藥典及各國藥典的升版,對阿米卡星的質量要求越來越高。在2020年版《中國藥典》中阿米卡星的雜質包含:雜質A、雜質B、雜質C、雜質D、雜質E、雜質F、雜質G、雜質H、雜質I等,雜質種類繁多,對雜質控制要求較高。阿米卡星作為本公司的拳頭產品,在國內外市場均有銷售,產量大、效益好,占有較大的市場份額,對于雜質的研究就非常有必要。通過雜質研究能夠對產品帶來較大的幫助,一方面可以減少雜質產生,提升質量,另一方面,反應過程雜質合成減少以后又能增加收率,降低生產成本。本次我們著重對雜質E進行研究,試圖找到雜質E的形成條件并進行雜質控制。
1 檢測方法的建立
1.1 衍生化后的HPLC法
衍生化處理過程中使用到的衍生劑2,4,6-三硝基苯磺酸的實驗操作安全性影響較大;衍生化法干擾因素多,如不同廠家的試劑有明顯差異,致使部分雜質的結果重現性差,不利于產品質量的全面控制,且衍生需要的時間較長。
1.2 薄層層析(TCL)方法
工藝中控方法采用TCL進行監控,雜質的分離度不夠,所監控的雜質有限,不能監控工藝中所涉及的大量雜質,中間生產過程不能有效的進行監控。為了改變目前中控手段的短板,擬對藥典方法進行借鑒,建立中控檢測方法。目前在使用的阿米卡星藥典方法有歐洲藥典電化學方法和中國藥典末端吸收方法。但由于歐洲藥典的有關物質檢測方法采用電化學檢測器,該檢測器使用金電極,金電極比較脆弱,一般適合于純度較高的成品檢測,不適合對產品的中間控制。故最終采用中國藥典的末端吸收方法來進行檢測與有關物質的控制,該法具有檢測樣品處理簡單、檢測時間短、專屬性強、靈敏度高、重復性好的優點。
分析方法的建立:
供試品溶液:取本品適量,加流動相A溶解并稀釋制成每1 mL中約含5 000 u單位的溶液,作為供試品溶液;
對照溶液:精密量取適量,用流動相A稀釋制成每1 mL中約含0.05 mg的溶液,作為對照溶液;
色譜條件:按照本公司高效液相色譜法標準操作規程試驗,引用2020年版中國藥典二部的規定[1]。
按下式進行計算:
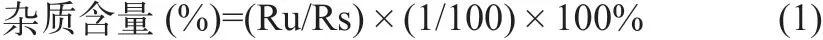
式中:Ru為供試品峰面積;Rs為對照品峰面積。
并對專屬性、精密度、定量限、檢測限等方面進行了確認。
在完成過程檢測方法優化后,著重對阿米卡星雜質E進行了研究。
2 阿米卡星雜質E形成條件探討及合成條件控制
2.1 阿米卡星雜質E形成條件
目前阿米卡星生產工藝常采用卡那霉素經過硅烷化保護氨基后與PHBA活性酯酰化來制備,再經過水解、肼解、過柱、結晶得到。其中硅烷化的合成路線如圖2所示下[2]。
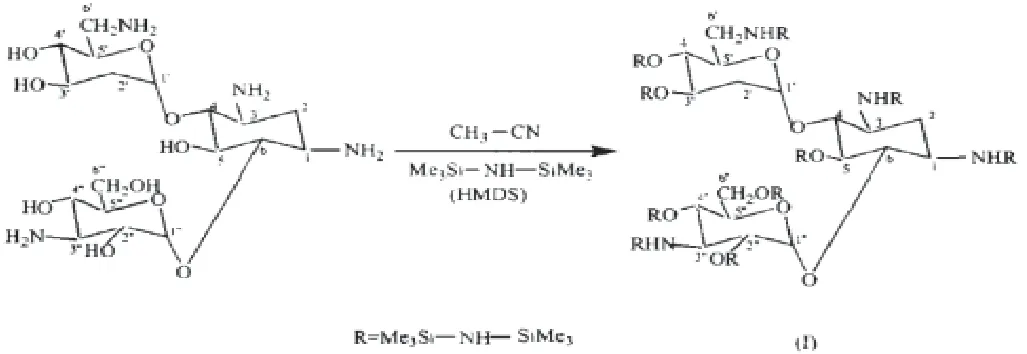
圖2 硅烷化的合成路線
雜質E為阿米卡星的雜質之一,根據藥典雜質結構分析為卡那霉素的6′位的氨基加上了AHBA側鏈。而卡那霉素分子中有4個氨基分別為1,3,6′,3″四個位置的氨基,每個氨基都可參與反應,這些氨基未進行保護而直接與活性酯反應時,6′位的氨基反應活性最大,但這不是需要的產物。因此需要改變氨基的活性,讓1位的活性最大,最大量的生成阿米卡星。因此通過硅烷化保護反應后,1位上的氨基活性最強,6′位變成最弱,合成產物以阿米卡星為主。對生產過程中的肼解后濾液進行了檢測,發現雜質E含量平均達到了3.5%,雜質水平偏高,通過前述合成路線分析認為硅烷化保護程度影響雜質水平。為此,我們將通過開展一系列的實驗進行摸索確認影響雜質E的合成條件,并找到最佳反應條件進行雜質控制。
2.2 雜質合成條件控制
合成路線:將一定量的卡那霉素投入無水乙腈中,加入六甲基二硅烷胺,再加入三甲基后升溫,到達回流溫度,回流一定的時間后結束降溫,再經過酰化、水解、肼解。從合成路線中我們分析得到兩個主要影響因素,六甲基二硅烷胺的量及回流反應時間,這兩個因素影響著硅烷化對氨基的保護程度,下面就這兩個因素開展以下實驗:
2.2.1 實驗部分
(1)六甲基二硅烷胺的使用量
在四口燒瓶加入300 mL無水乙腈,投入46.0 g卡那霉素(分子量為484.5,94.94 mmol),加入六甲基二硅烷胺(分子量為161.39),再加入10 g三甲基氯硅烷后升溫,到達回流溫度后回流1.5 h。針對六甲基的使用量,設置了摩爾比1∶0、1∶2、1∶5.5、1∶6.5、1∶7.5等5種情況,做到肼解后濾液(上柱分離前中間體)結果如表1所示。
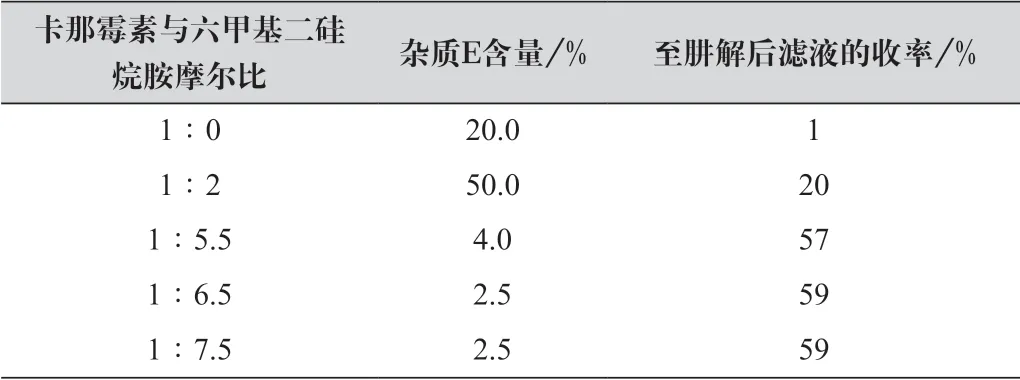
表1 六甲基二硅烷胺量的影響
從表中可以看出卡那霉素與六甲基二硅烷胺摩爾比1∶6.5時,雜質E的含量已經最低,是最佳條件之一。把這個因素固定后,又對回流反應時間開展實驗。
(2)硅烷化回流反應時間
設置了回流時間2、4、6、8 h四種情況,做到肼解后濾液(上柱分離前中間體),結果如表2所示。
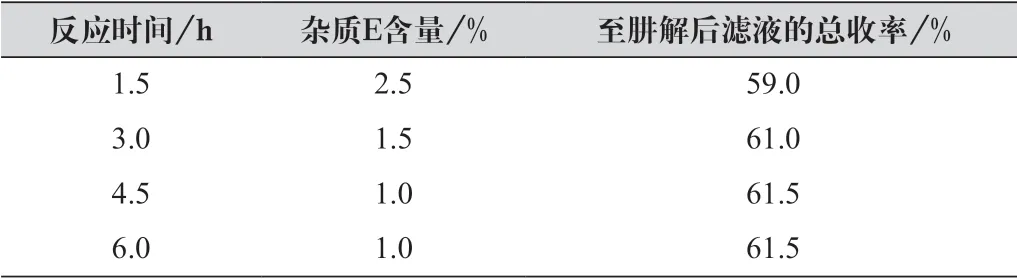
表2 硅烷化回流反應時間的影響
從表2可以看出回流時間為4.5 h時,雜質E的含量最低,是最佳條件之一。
2.2.2 合成控制小結
通過六甲基二硅烷胺的使用量量及硅烷化反應時間兩個條件的探索,發現卡那霉素與六甲基摩爾比在1∶6.5,反應時間在4.5 h為最佳,雜質E的含量最低在1%左右,而我們的收率也最高,達到了61.5%,收率提升了2.5%左右。
2.3 雜質E的柱層析控制
通過柱層析分離對合成料液進行分離,達到純化的目的。柱層析是一個雜質分離過程,主要是通過樹脂上柱吸附和解析劑洗脫達到分離的效果來去除料液中的未反應的卡那霉素及其他雜質如雜質E。由于柱層析的周期長,對過程的控制尤為重要。建立前述的中控檢測方法以后,對雜質E的解析過程進行了有效監控,雜質E得到有效控制。通過這樣的控制雜質E控制在0.1%以內,達到了較好的分離效果。
3 結語
本次研究首先建立了阿米卡星中間體的中控方法,該方法具有樣品處理簡單、檢測時間短、專屬性強、靈敏度高、重復性好的優點。其次對雜質E形成條件進行探討,并利用中控方法,對合成條件進行研究,確立了最佳的合成條件。再次對柱層析過程加強控制,控制雜質E在0.1%以內。通過本次研究,一方面提升了合成步驟的收率,提升2.5%,另一方面有效降低了雜質E的含量,為柱層析減輕壓力,保證了產品質量,取得了比較理想的成果。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
