QuEChERS結合高效液相色譜串聯質譜測定枇杷中氯苯嘧啶醇殘留量
2021-09-22 22:16:24胡彧嫻林薌華賴添財
福建農業科技 2021年7期
胡彧嫻 林薌華 賴添財


摘 要:建立了QuEChERS結合高效液相色譜串聯質譜測定枇杷中氯苯嘧啶醇殘留量方法,樣品經乙腈提取,N丙基乙二胺(PSA)凈化,高效液相色譜串聯質譜測定。結果表明:氯苯嘧啶醇在0.5~200 μg·L-1濃度范圍內線性關系良好( r 2=0.999 4),檢出限為0.14 μg·kg-1,定量限為0.5 μg·kg-1;氯苯嘧啶醇在2、20、200 μg·kg-1添加水平下,平均回收率分別為87.0%、97.4%和96.7%,相對標準偏差(RSD)分別為8.3%、7.9% 和4.3%。該方法簡單快速,靈敏度高,能夠滿足殘留檢測要求。
關鍵詞:氯苯嘧啶醇;枇杷;殘留;QuEChERS法;高效液相色譜串聯質譜
中圖分類號:S 481.8?? 文獻標志碼:A?? 文章編號:0253-2301(2021)07-0069-05
DOI: 10.13651/j.cnki.fjnykj.2021.07.013
Determination of Fenarimol Residues in Loquat by UsingQuEChERS Combined with HPLCMS/MS
HU Yuxian, LIN Xianghua, LAI Tiancai*
(Zhangzhou Agricultural Inspection and Monitoring Center, Zhangzhou, Fujian 363000, China)
Abstract: A method based on QuEChERS combined with high performance liquid chromatographytandem mass spectrometry (HPLCMS/MS) was established for the determination of fenarimol residues in loquat. The samples were extracted by acetonitrile, purified by Nprimary secondary amine (PSA), and determined by high performance liquid chromatographytandem mass spectrometry. The results showed that: the linearity of fenarimol was good in the concentration range of 0.5~200 μg·L-1 ( r 2=0.999 4), the limit of detection was 0.14 μg·kg-1, and the limit of quantitation was 0.5 μg·kg-1. The average recovery rates of fenarimol were respectively 87.0%, 97.4% and 96.7% at the adding level of 2, 20 and 200 μg·kg-1, with the relative standard deviations (RSD) of 8.3%, 7.9% and 4.3%, respectively. The method was simple, rapid and sensitive, which could meet the requirements of residue detection.
Key words: Fenarimol; Loquat; Residue; Method of QuEChERS; HPLCMS/MS
氯苯嘧啶醇又名樂必耕,異嘧菌醇,屬低毒、廣譜的內吸性殺菌劑,兼有保護和治療作用,是一種麥角甾醇生物合成抑制劑,能抑制病原菌菌絲生長[1-3],主要應用于果樹、蔬菜及觀賞植物等,對白粉病、黑星病、銹病等有一定的防治效果[3-4]。枇杷果肉營養豐富,風味佳良,具有止咳潤肺、下氣等功效[5],深受人們的喜愛,在福建、廣東、廣西、臺灣等地廣泛種植,而漳州市是福建省枇杷種植的主要產區之一。在栽培過程中為防治枇杷病蟲害,農藥的使用成為農戶果園管理的重要手段。
迄今,國內對氯苯嘧啶醇的研究主要集中在光化學降解與環境轉歸方面[6-10],對其殘留檢測方法的研究較少[11-13],而在枇杷上的殘留檢測方法尚未見報道。農產品中農藥殘留量的檢測,其樣品前處理方法主要有固相萃取法、液液萃取法、凝膠滲透色譜(GPC)法和QuEChERS法等,QuEChERS(quick,easy,cheap,effective,rugged and safe)法是由Anastassiades等[14]于2003年提出,QuEChERS前處理方法相較于其他方法,操作簡單、靈敏度高,已在農藥殘留、獸藥殘留等領域得到驗證和推廣。為此,本研究采用QuEChERS進行樣品前處理,高效液相色譜串聯質譜測定枇杷中氯苯嘧啶醇殘留量,建立QuEChERS結合高效液相色譜串聯質譜測定方法,旨在為監測枇杷中氯苯嘧啶醇的殘留量提供依據。
1 材料與方法
1.1 供試材料
1.1.1 試劑 氯苯嘧啶醇標準品(農業部環境保護科研監測所,100 mg·L-1);無水硫酸鎂、氯化鈉(西隴科學股份有限公司,分析純);N丙基乙二胺
[(PSA),德國CNW公司];乙腈、甲醇、丙酮(美國天地試劑公司,色譜純);甲酸(山東西亞化學工業有限公司,色譜純);試驗用水為超純水。
1.1.2 主要儀器設備 DIONEX Ultimate 3000Thermo TSQ Quantum Ultra EMR高效液相色譜串聯質譜儀(美國ThermoFisher公司,帶ESI源);318K離心機(德國Sigma公司) ;QUINTIX612上皿式天平(德國賽多利斯科學儀器有限公司)。
1.2 試驗方法
1.2.1 標準溶液配制 準確吸取0.5 mL氯苯嘧啶醇標準品(100 mg·L-1)于10 mL容量瓶中,用乙腈稀釋定容,配制成5 mg·L-1的標準儲備液,于0~4℃冰箱中避光保存備用。根據需要分別用乙腈或空白基質稀釋成適當濃度的標準工作溶液,現配現用。
1.2.2 樣品提取與凈化 參考SN/T 4138-2015《出口水果和蔬菜中敵敵畏、四氯硝基苯、丙線磷等88種農藥殘留的篩選檢測QuEChERS氣相色譜負化學源質譜法》[15]并適當改進,稱取10 g(精確至0.01 g)枇杷樣品置于50 mL離心管中,準確加入20 mL乙腈、4 g無水硫酸鎂和1 g氯化鈉,渦旋混合2 min,于4 500 r·min-1的離心機中離心5 min。取上層有機相6.0 mL轉移至裝有0.30 g PSA、0.90 g無水硫酸鎂的離心管中,渦旋振蕩1 min,取1 mL過0.22 μm有機濾膜,待高效液相色譜串聯質譜儀分析測定。
1.2.3 檢測條件 (1)液相色譜條件。色譜柱為Atlantis T3 Column(100 mm×2.1 mm×3 μm);流動相A為0.1%甲酸水溶液,流動相B為乙腈;進樣量為2.0 μL;流速為0.25 mL·min-1;總運行時間為10 min;梯度洗脫程序見表1。(2)質譜條件。電噴霧正離子掃描(ESI+);噴霧溫度為260℃;噴霧電壓為3 900 V;離子傳輸管溫度為300℃;鞘氣(N2)流速為3.58 L·min-1,輔助氣(N2) 流速為5.74 L·min-1,掃描模式:SRM。
1.2.4 提取溶劑的選擇 分別采用甲醇、丙酮、乙腈作為提取溶劑,其余操作步驟均按1.2.2的樣品提取與凈化進行,添加氯苯嘧啶醇標準溶液進行提取回收率試驗,根據提取效果確定最佳提取溶劑。
1.2.5 方法有效性評價 準確吸取5 mg·L-1的標準儲備液稀釋,配制成0.5、1、5、10、50、100、200 μg·L-1的基質匹配標準工作溶液。以質量濃度為橫坐標、對應的峰面積為縱坐標繪制標準曲線,評價方法的線性范圍和靈敏度,外標法定量。在枇杷空白樣品中添加3個水平的標準工作溶液進行添加回收試驗,每個水平重復5次,按1.2.2方法進行操作,測定添加回收率和相對標準偏差(RSD),評價方法的準確度和精密度。
2 結果與分析
2.1 質譜條件的優化
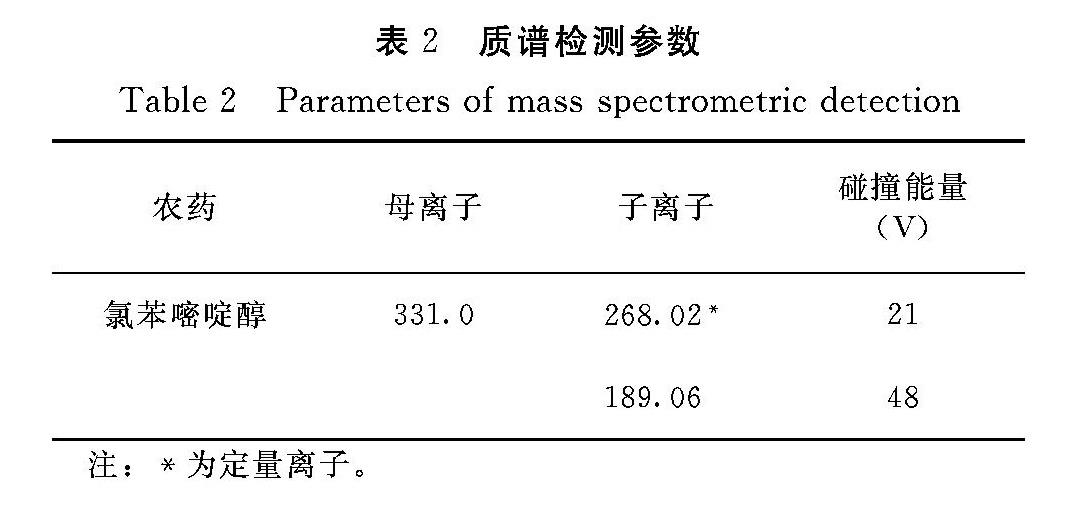
試驗采用0.1%甲酸水溶液和乙腈作為流動相,將氯苯嘧啶醇標準品稀釋到1 mg·L-1,經注射泵注入質譜,選擇ESI+模式,調節合適的注射泵流速及噴霧溫度、電壓等條件使響應值信號大于106,且響應達到穩定狀態。先全掃描確定母離子,再選擇反應監測模式(SRM)進行二級質譜自動優化,得到定性和定量離子及相應的碰撞能量,最終確定氯苯嘧啶醇的質譜檢測參數(表2)。
2.2 提取溶劑的確定
農藥殘留檢測前處理常用的提取劑較多,本試驗考察了甲醇、丙酮、乙腈3種溶劑的提取效果,結果表明氯苯嘧啶醇(添加量為100 μg·kg-1)在這3種溶劑中的平均回收率分別77.2%、97.4%和98.6%,即丙酮、乙腈的提取效果優于甲醇。盡管在丙酮和乙腈溶劑中的回收率相差不大,但丙酮較易提取出其他雜質,為此,本試驗優選乙腈作為提取劑。
2.3 方法的線性范圍和靈敏度
由于樣品中基質效應的存在,本試驗采用基質匹配標準溶液來進行校正。通過向空白基質中添加不同濃度的標樣,配制基質匹配系列標準工作溶液并進樣分析,以標準工作溶液的質量濃度為橫坐標、對應的峰面積為縱坐標,在0.5~200 μg·L-1濃度范圍內繪制標準曲線(圖1)。氯苯嘧啶醇的線性回歸方程為 y=14 799.6x-5 369.26(r2=0.999 4), 線性關系良好。以信噪比(S/N)的3倍和10倍計算檢出限和定量限,氯苯嘧啶醇檢出限為0.14 μg·kg-1、定量限為0.5 μg·kg-1,可滿足定量分析的要求。
2.4 方法的準確度和精密度
在枇杷空白樣品中分別加入2、20和200 μg·kg-1 3個質量水平的標準工作溶液進行添加回收試驗,重復5次。按上述方法進行前處理和上機測定,枇杷空白樣品和加標樣品的提取離子流色譜圖見圖2和圖3,氯苯嘧啶醇保留時間為6.21 min;氯苯嘧啶醇的平均回收率為87.0%~97.4%、RSD為4.3%~8.3%(表3),表明該方法適用于枇杷中氯苯嘧啶醇的殘留測定。
3 結論與討論
本試驗中供試的甲醇、丙酮、乙腈3種提取劑,甲醇的提取效果呈現最差,這可能與其較難與水層分離有關[16]。農產品中農藥殘留量的檢測前處理,常見的凈化劑有PSA、C18、GCB等,在試驗中可以根據樣品的特性來選擇合適的凈化劑。PSA主要用于除去基質中極性物質如糖類、有機酸、脂肪酸及親脂性色素等極性物質[17];C18可以去除脂肪酸、脂類等非極性干擾物[18];GCB對色素有較強的吸附凈化能力,但會吸附一些平面及對稱結構的農藥,影響農藥的回收率[19]。枇杷樣品提取液顏色極淺,色素、脂類等干擾物較少,為此,本試驗中在凈化時加入PSA就能達到較好凈化效果。
關于農產品中氯苯嘧啶醇殘留檢測方法的研究,王姍姍等[13]建立的QuEchERS超高效液相色譜串聯質譜法,測定草莓中氯苯嘧啶醇的檢出限為0.5 μg·kg-1;梁睿等[12]建立的蘋果中氯苯嘧啶醇殘留檢測方法,測定枇杷中氯苯嘧啶醇的檢出限為<0.3 μg·kg-1。本試驗建立的QuEChERS結合高效液相色譜-串聯質譜檢測方法,對枇杷中氯苯嘧啶醇的檢出限可達0.14 μg·kg-1、定量限為0.5 μg·kg-1,平均回收率為87.0%~97.4%、RSD為4.3%~8.3%。對枇杷中氯苯嘧啶醇的檢出限可達0.14 μg·kg-1、定量限為0.5 μg·kg-1,平均回收率為87.0%~97.4%、RSD為4.3%~8.3%,符合NY/T 788-2018《農作物中農藥殘留試驗準則》[20]規定的要求。同時,根據GB 2763-2019《食品安全國家標準食品中農藥最大殘留限量》[21]規定,枇杷中氯苯嘧啶醇的MRL為0.3 mg·kg-1,能滿足于GB 2763-2019的要求。由上表明,本試驗所建立的枇杷中氯苯嘧啶醇殘留量分析方法,樣品經乙腈提取、N丙基乙二胺(PSA)凈化、高效液相色譜串聯質譜測定,方法簡單快速、靈敏度高,能夠滿足殘留檢測要求。
參考文獻:
[1]王寧.氯苯嘧啶醇的合成[J].現代農藥,2005,4(3):17-18.
[2]王丹軍,岳永德,湯鋒,等.7種農藥和5種表面活性劑對氯苯嘧啶醇光解的影響[J].安徽農業大學學報,2007,34(1):45-48.
[3]王丹軍,岳永德,湯鋒,等.殺菌劑氯苯嘧啶醇的環境轉歸研究進展[J].安徽農業大學學報,2008,36(22):9699-9701.
[4]孫家隆,齊軍山.現代農藥應用技術叢書.殺菌劑卷[M].北京:化學工業出版社,2015:123-124.
[5]魏云瀟,何良興.塘棲枇杷重金屬和農藥殘留及套袋對品質的影響[J].浙江農業科學,2012,(4):513-514.
[6]王丹軍,岳永德,湯鋒,等.幾種表面活性劑和色素物質對氯苯嘧啶醇光化學降解的影響[J].安徽農業大學學報,2009,37(3):1294-1297,1306.
[7]王丹軍,岳永德,湯鋒,等.氯苯嘧啶醇在水體中的光解機制研究[J].環境污染與防治,2008,30(7):47-51.
[8]王丹軍,岳永德,湯鋒,等.氯苯嘧啶醇在有機溶劑中的光化學降解研究[J].安徽農業大學學報,2007,34(1):40-44.
[9]王丹軍,岳永德,湯鋒,等.陰離子對氯苯嘧啶醇光化學降解的影響[J].農業環境科學學報,2008,27(5):2023-2027.
[10] JACKSON R,LEWIS C.The metabolism of 14Cfenarimol soil[J]. Dow Elanco Reference, 1994(15):32-45.
[11]梁睿,岳永德,湯鋒.氯苯嘧啶醇在蘋果中殘留檢測方法的研究[J].中國農學通報,2010,26(12):57-62.
[12]梁睿,岳永德,湯鋒.蘋果中氯苯嘧啶醇殘留檢測方法的質量控制研究[J].安徽農業科學,2006,34(20):5157-5158,5188.
[13]王姍姍,王莉,程媛媛,等.QuEchERS超高效液相色譜串聯質譜法同時測定草莓中85種農藥殘留[J].浙江農業科學,2018,59(9):1584-1591.
[14]ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and ″dispersive solidphase extraction″ for the determination of pesticide residues in produce[J]. Journal of AOAO International, 2003,86(2):412-431.
[15]中華人民共和國國家質量監督檢驗檢疫局.SN/T 4138-2015出口水果和蔬菜中敵敵畏、四氯硝基笨、丙線磷等88種農藥殘留的篩選檢測QuEChERS氣相色譜負化學源質譜法[S].北京:中國標準出版社,2015:2.
[16]戴平望.超高效液相色譜串聯質譜法測定茶葉中5種農藥殘留量[J].理化檢驗-化學分析,2014,50(4):508-511.
[17]李萍萍,張月,吳小芳,等.固相萃取法與QuEChERS法對比檢測無核荔枝中19種農藥殘留[J].熱帶作物學報,2017,38(8):1565-1571.
[18]高陽,徐應明,孫揚,等.QuEChERS提取法在農產品農藥殘留檢測中的應用進展[J].農業資源與環境學報,2014,31(2):110-117.
[19]李蓉,儲大可,張朋杰,等.QuEChERS/HPLCMS/MS法測定黃瓜、菜心、葡萄、香蕉中127種農藥殘留[J].分析測試學報,2015,34(5):502-511.
[20]中華人民共和國農業農村部.NY/T 788-2018農作物中農藥殘留試驗準則[S].北京:中國農業出版社,2018:5.
[21]中華人民共和國國家衛生健康委員會,中華人民共和國農業農村部,國家市場監督管理總局.GB 2763-2019食品安全國家標準食品中農藥最大殘留限量[S].北京:中國農業出版社,2019:162.
(責任編輯:柯文輝)
