鋰硫電池中的催化應(yīng)用
2021-09-28 03:35:58高希雅鄧子華李存璞魏子棟
化工進(jìn)展 2021年9期
高希雅,鄧子華,李存璞,魏子棟
(重慶大學(xué)化學(xué)化工學(xué)院,重慶 400044)
1 鋰硫電池基本原理與存在問(wèn)題
可持續(xù)能源技術(shù)的蓬勃發(fā)展標(biāo)志著現(xiàn)代社會(huì)的能源革命,探索可持續(xù)、清潔、安全的新能源供應(yīng)已成為了國(guó)際共識(shí)。新能源電動(dòng)汽車、便攜式電子設(shè)備和移動(dòng)電源等領(lǐng)域的興起使得人們對(duì)儲(chǔ)能設(shè)備提出了更高的要求。在熱能、風(fēng)能、機(jī)械能等多種儲(chǔ)能系統(tǒng)中,具有高能量密度儲(chǔ)能的可充電電池成為了最常用的電化學(xué)儲(chǔ)能設(shè)備。其中鋰硫電池以其高理論容量(1672mA·h/g)、豐富的自然資源和環(huán)境友好性等優(yōu)點(diǎn)受到越來(lái)越多的關(guān)注,是新興電池技術(shù)中極具吸引力和發(fā)展前景的候選技術(shù),是滿足全球日益增長(zhǎng)的能源消費(fèi)需求的理想下一代儲(chǔ)能設(shè)備。
鋰硫電池的充放電過(guò)程有溶解和沉積兩個(gè)步驟,包含固相-液相-固相的相變轉(zhuǎn)化。在放電過(guò)程中,正極的單質(zhì)S8被多步還原成Li2S,放電曲線通常會(huì)出現(xiàn)兩個(gè)平臺(tái),分別位于2.4V和2.1V左右,如圖1所示。第Ⅰ階段的放電平臺(tái)為2.4V,對(duì)應(yīng)單質(zhì)S8開(kāi)環(huán)并與遷移到正極的鋰離子成鍵生成Li2S8的固-液兩相還原過(guò)程和Li2S8向可溶性長(zhǎng)鏈Li2S6轉(zhuǎn)變的液-液?jiǎn)蜗噙€原過(guò)程;第Ⅱ階段的電壓從2.4V驟降為2.1V,可溶性Li2S6再還原生成可溶性Li2S4,且第Ⅰ、Ⅱ階段共提供約25%的理論硫容量。第Ⅲ階段的放電平臺(tái)為2.1V,包含可溶性Li2S4向不溶的短鏈Li2S2轉(zhuǎn)變的液-固兩相還原過(guò)程和Li2S2向Li2S 轉(zhuǎn)變的固-固單相還原過(guò)程,貢獻(xiàn)約75%的理論硫容量[1]。在充電過(guò)程中,Li2S 首先轉(zhuǎn)化為不溶的短鏈Li2S2,然后進(jìn)一步生成可溶性長(zhǎng)鏈Li2Sn(4≤n≤8,n為整數(shù))(LiPSs),最后轉(zhuǎn)化為S8。電極反應(yīng)和電池總反應(yīng)如式(1)~式(3)。

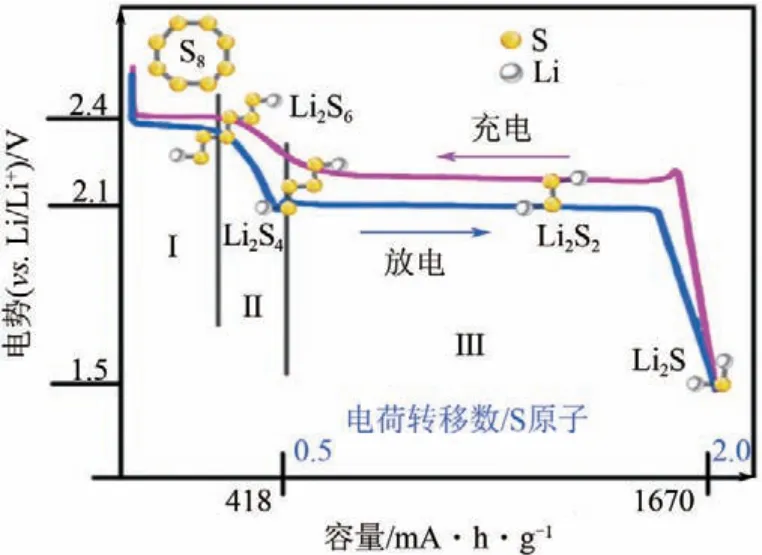
圖1 鋰硫電池的充放電曲線[1]
鋰硫電池作為具有應(yīng)用潛力的高能量密度儲(chǔ)能系統(tǒng),當(dāng)前仍存在眾多挑戰(zhàn),嚴(yán)重影響著電池的倍率性能和循環(huán)壽命,使其不能達(dá)到預(yù)期的理論水平,極大地阻礙了實(shí)際商業(yè)應(yīng)用化進(jìn)程[2-3]。具體問(wèn)題如下:①單質(zhì)S8和固體產(chǎn)物L(fēng)i2S2/Li2S的導(dǎo)電性較差,限制了電子在硫陰極中的傳遞和活性硫的利用;②穿梭效應(yīng)造成活性物質(zhì)硫的不可逆損失,使得電池自放電現(xiàn)象嚴(yán)重,導(dǎo)致電池庫(kù)侖效率降低,循環(huán)壽命縮短[4];③充放電過(guò)程中硫的體積膨脹,導(dǎo)致宿主材料的結(jié)構(gòu)易粉粹和崩塌,影響電池的循環(huán)性能。
以上問(wèn)題中,由不同濃度梯度的LiPSs 造成的穿梭效應(yīng)最能影響鋰硫電池的性能。在實(shí)際放電反應(yīng)進(jìn)程中,可溶性LiPSs 和不溶性Li2S2/Li2S 之間存在相的轉(zhuǎn)化,且固體Li2S2的成核勢(shì)壘較大,其成長(zhǎng)速度大于成核速度,導(dǎo)致固體產(chǎn)物生成滯后,進(jìn)一步導(dǎo)致可溶性LiPSs 在正極側(cè)的堆積過(guò)剩。反觀由于Li2S 具有較高的活化能和較差的導(dǎo)電性,使得Li2S在充電過(guò)程中難以完全再氧化,不均勻滯留在正極表面,降低了反應(yīng)的可逆性和可循環(huán)性。因此,研究者們開(kāi)始關(guān)注如何減緩穿梭效應(yīng)的同時(shí)提高反應(yīng)動(dòng)力學(xué),從而加快整個(gè)反應(yīng)進(jìn)程,提出了鋰硫電池中的催化作用,認(rèn)為推動(dòng)可溶性LiPSs 和固態(tài)Li2S2/Li2S的快速互相轉(zhuǎn)化,克服液固轉(zhuǎn)化中的能壘是實(shí)現(xiàn)鋰硫電池高能量密度的一條有希望的途徑[5]。
2 鋰硫電池催化材料
與可溶性LiPSs 的物理約束相比,化學(xué)吸附和催化轉(zhuǎn)化表現(xiàn)出更好的抑制穿梭效應(yīng)的能力[6-7]。本節(jié)總結(jié)了各種應(yīng)用于化學(xué)吸附和催化轉(zhuǎn)化可溶性LiPSs 的材料,包括過(guò)渡金屬氧化物、氮化物、硫化物、碳化物、磷化物、金屬有機(jī)框架、共價(jià)有機(jī)框架、導(dǎo)電性載體等。
2.1 過(guò)渡金屬氧化物
過(guò)渡金屬化合物(MaXb,M 為金屬,X 為陰離子)是一種用途廣泛的材料,因?yàn)樗鼈兊奈锢?化學(xué)性質(zhì)取決于M和X。大多數(shù)過(guò)渡金屬化合物具有適合與硫類化學(xué)反應(yīng)的極性表面,因此它們占鋰硫電池催化材料中的大部分[8]。過(guò)渡金屬氧化物具有較大的電化學(xué)活性表面,并含有親水性基團(tuán),是很好的硫宿主材料[8]。O 原子具有空d 軌道,是良好的電子接受體,容易和鋰或其他金屬化學(xué)鍵合,提高與可溶性LiPSs 的結(jié)合能,相互作用強(qiáng)烈,降低了Li2S 成核的動(dòng)力學(xué)能壘,有利于Li2S 的沉積和分解。
王曉敏團(tuán)隊(duì)[9]提出以Fe3O4納米粒子/分級(jí)多孔碳(Fe3O4/HPC)為陰極和FeP/HPC 改性隔膜的雙功能策略,以提高LiPSs 的錨定和催化性能,保證均勻的Li2S沉積,降低死硫。結(jié)果表明,組裝電池在1C的電流密度下循環(huán)1000次后,以0.083%極低的衰減率使得容量保持為562.4mA·h/g。系統(tǒng)的理論計(jì)算如圖2 所示,由于Fe—S 鍵和Li—O 鍵的存在,p帶中心發(fā)生了偏移,使得Fe3O4與Li2S4和Li2S6的結(jié)合能更強(qiáng),加速了Li2S4和Li2S6的氧化還原動(dòng)力學(xué)反應(yīng),提高了固體Li2S沉淀的均勻成核生長(zhǎng)。
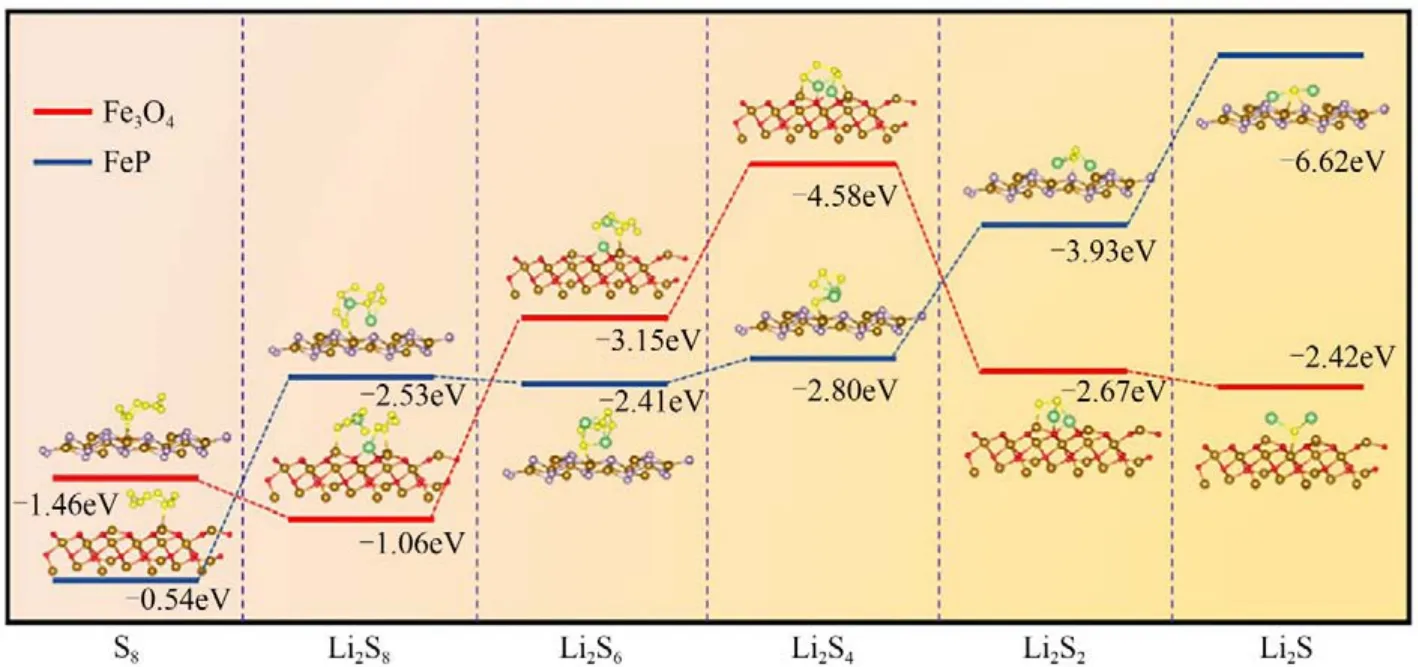
圖2 不同反應(yīng)產(chǎn)物的DFT計(jì)算模型和吸附能[9]
Nazar 等[10]用MnO2殼層包裹硫顆粒,利用KMnO4與硫之間的氧化還原反應(yīng)。形成的納米級(jí)MnO2殼層與可溶性LiPSs 在物理和化學(xué)上相互作用,并有效捕獲它們,這使得1700 多次循環(huán)后還能保持0.039%的低容量衰減。Lu 等[11]研究了將單質(zhì)硫固定在G/MnO2復(fù)合材料表面以提高硫電極的循環(huán)穩(wěn)定性的方法。由于多孔石墨烯具有良好的導(dǎo)電性和MnO2的催化性能,電極為G/MnO2/S 的電池在0.2C和1C下的初始放電容量分別為1132mA·h/g和908mA·h/g。1C下完成500個(gè)循環(huán)后的保留率為43.78%以上。
Liu等[12]和Nazar等[13]分別證明MnO2和V2O5可以將LiPSs 氧化到高氧化態(tài)以達(dá)到限制中間產(chǎn)物的作用。高氧化還原電位的金屬氧化物可以與LiPSs 相互作用形成硫代硫酸鹽/聚硫酸鹽表面物種,甚至通過(guò)表面氧化還原反應(yīng)形成硫酸鹽物種,從而抑制LiPSs 的遷移,提供鋰硫電池的循環(huán)穩(wěn)定性[14]。例如,LiPSs與MnO2之間的氧化還原反應(yīng)可以發(fā)生在MnO2的表面[13]。隨著Mn4+還原為Mn2+,MnO2表面的多硫離子被氧化,生成硫代硫酸鹽基團(tuán)。表面的硫代硫酸鹽基團(tuán)作為錨定位點(diǎn),使LiPSs 形成聚硫酸鹽并引發(fā)Li2S2/Li2S的轉(zhuǎn)化。
CeO2[15]、TiO2[16]、WO3/ZrO2[17]等多種氧化物催化LiPSs 的轉(zhuǎn)化均有報(bào)道。此外,雙金屬氧化物如Ni-Fe等[18]具有多催化位點(diǎn)而受到關(guān)注。
2.2 過(guò)渡金屬氮化物
金屬氮化物對(duì)LiPSs 的轉(zhuǎn)化具有很強(qiáng)的催化作用,其高電導(dǎo)率是主要優(yōu)勢(shì),它可以使活性硫快速傳遞,從而提高活性硫的電化學(xué)利用率。
李紅團(tuán)隊(duì)[19]通過(guò)構(gòu)建有氮化鈷(Co4N)催化劑的雙殼中空納米籠提高了LiPSs氧化還原動(dòng)力學(xué)。N摻雜的中空內(nèi)碳?xì)げ粌H可以作為L(zhǎng)iPSs 的物理化學(xué)吸收體,而且提高了電極的電導(dǎo)率,顯著抑制了穿梭效應(yīng)。Co4N 納米粒子嵌入在N 摻雜碳的外殼中,催化LiPSs 的轉(zhuǎn)化,在循環(huán)過(guò)程中極化減少,動(dòng)力學(xué)加快。鋰離子插層能量學(xué)的理論研究證實(shí)了與金屬Co 催化劑相比,Co4N 催化劑的催化活性得到了提高。結(jié)果表明,該電極在5C時(shí)循環(huán)400次后容量保持為658mA·h/g,具有良好的穩(wěn)定性。
Zheng等[20]合成了一種Fe2N@C納米盒(Fe2N@CNBs)多功能硫主體,通過(guò)蝕刻與氮化結(jié)合的策略,用于高速率和長(zhǎng)循環(huán)的鋰硫電池。高導(dǎo)電性的碳?xì)の锢砩舷拗屏嘶钚圆牧希⑻峁┝丝焖匐娮?離子傳輸?shù)挠行緩健M瑫r(shí),密度泛函理論計(jì)算和電化學(xué)分析證明了極性Fe2N核對(duì)LiPSs具有較強(qiáng)的化學(xué)鍵合和有效的催化活性。S/Fe2N@CNBs 電極即使在1C下循環(huán)600 次后,其容量仍保持在881mA·h/g,平均衰減率僅為0.036%,表現(xiàn)出高比容量、優(yōu)越的倍率能力和長(zhǎng)期循環(huán)穩(wěn)定性。
氮化鈦(TiN)表面與硫之間存在極強(qiáng)的鍵合,使得TiN 表面同時(shí)具有催化LiPSs 轉(zhuǎn)化的活性和顯著的錨定效應(yīng)[21]。此外,TiN 有助于克服LiPS化學(xué)歧化反應(yīng)遲緩的動(dòng)力學(xué)問(wèn)題。總的來(lái)說(shuō),TiN大大提高了鋰硫電池的容量和速率能力,在5C下放電容量甚至達(dá)到700mA·h/g。除TiN 外,高導(dǎo)電性的InN[22]、VN[23]等也具有類似的電催化活性,被用于加快LiPSs轉(zhuǎn)化反應(yīng)。
2.3 過(guò)渡金屬硫化物
近年過(guò)渡金屬硫化物因其化學(xué)吸附能力和電催化作用而被發(fā)展成為理想的主體材料[24]。具有特殊離域電子結(jié)構(gòu)的硫化物削弱了金屬-硫鍵的離子性質(zhì),相比O2-來(lái)說(shuō),S2-是更軟的堿,因而導(dǎo)電性得到了提升[25]。此外,硫化物的極性性質(zhì)可以誘導(dǎo)LiPSs的快速轉(zhuǎn)化,產(chǎn)生更多的活性位點(diǎn)[26-28],并促進(jìn)Li2S的沉積行為,具有高催化效率的優(yōu)點(diǎn),進(jìn)而提高活性硫的利用率,促進(jìn)鋰硫電池的反應(yīng)動(dòng)力學(xué)。
將納米結(jié)構(gòu)的VS4錨定在富含缺陷的碳納米纖維(CNF)上[26],生成CNF-VS4并被包覆在商業(yè)隔膜上,作為電催化劑以增強(qiáng)LiPSs 生成Li2S 的反應(yīng)動(dòng)力學(xué)。極性納米結(jié)構(gòu)的VS4對(duì)LiPSs表現(xiàn)出較高的親和力,導(dǎo)電的CNF網(wǎng)絡(luò)作為“第二捕集劑”對(duì)活性物質(zhì)進(jìn)行捕獲和再利用。功能涂層也為L(zhǎng)i2S的分散和穩(wěn)定提供了豐富的缺陷。當(dāng)CNF-VS4功能隔膜基于高硫含量(質(zhì)量分?jǐn)?shù)80%)陰極時(shí),在0.2C和2C 時(shí)的初始比容量也可以分別達(dá)到1135mA·h/g 和780mA·h/g。即使在更高的倍率5C 下,在經(jīng)過(guò)1000 次循環(huán)后仍然提供約為300mA·h/g 的穩(wěn)定容量。
Cao 等[27]利用原位合成的方法,通過(guò)CNTs 和ZIF-67 雜化結(jié)構(gòu)制備均勻分散的Co3S4催化納米顆粒,記為CNTs/Co3S4@NC。這種優(yōu)化的結(jié)構(gòu)使得硫和Co3S4納米顆粒在ZIF-67 衍生物的N 摻雜碳納米立方體內(nèi)能夠均勻分布和緊密接觸,從而與LiPSs化學(xué)鍵合產(chǎn)生有效的相互作用,增強(qiáng)鋰離子擴(kuò)散,達(dá)到最大的催化轉(zhuǎn)化效果,同時(shí)內(nèi)置的三維碳納米管網(wǎng)絡(luò)確保了電極的高導(dǎo)電性。即使在高倍率5C下,也能維持約850mA·h/g 的比容量,循環(huán)1000次后保持85%的穩(wěn)定性,對(duì)應(yīng)平均每循環(huán)容量衰減率僅為0.0137%。
孫學(xué)良課題組[28]提出了一個(gè)獨(dú)立結(jié)構(gòu)的核桃狀VS4納米位點(diǎn)結(jié)合CNTs 作為陰極。在這種框架下,CNTs 陣列為高硫負(fù)載提供了高表面積和導(dǎo)電性,而VS4納米位點(diǎn)有助于LiPSs 的捕獲和催化轉(zhuǎn)化。獨(dú)立CNTs 陣列和VS4納米位點(diǎn)具有高達(dá)6C 的高倍率能力,且VS4@CNTs/S 電極在2C 下以0.037%的低衰減率長(zhǎng)期循環(huán)1200次。
王連洲團(tuán)隊(duì)[29]首次報(bào)道了通過(guò)原位聚多巴胺輔助硫化過(guò)程,在N、S 共摻雜多孔碳(TiS2@NSC)約束下,Ti3C2TxMXene 成功轉(zhuǎn)化為三明治狀超薄TiS2納米片。作為硫的載體,即使在7.7mg/cm2高硫負(fù)載下,TiS2@NSC對(duì)LiPSs具有很高的捕獲能力和顯著的LiPSs還原和Li2S氧化的電催化活性。
崔屹課題組[30]對(duì)6 種金屬硫化物(FeS、SnS2、Ni3S2、VS2、TiS2和CoS2)進(jìn)行了研究。其中,VS2、TiS2和CoS2對(duì)Li2S 成核的動(dòng)力學(xué)勢(shì)壘較低,導(dǎo)致初始充電階段電位丘急劇減小,這歸因于鋰離子在這些金屬硫化物表面的高擴(kuò)散率,容易電荷轉(zhuǎn)移。因此,使用VS2、TiS2和CoS2的電極表現(xiàn)出穩(wěn)定的循環(huán)壽命和高容量。這些研究表明,即使沒(méi)有復(fù)雜的工程,金屬硫化物也具有出色的催化活性。
2.4 過(guò)渡金屬單原子催化材料
單原子催化是指穩(wěn)定在合適載體上的孤立金屬原子,可以提供高效和高活性的表面位置,從而可持續(xù)地催化轉(zhuǎn)化。單原子催化不僅最大限度地利用了活性中心,而且提高了效率,減少了金屬的使用,并提高了轉(zhuǎn)化過(guò)程中的催化選擇性。
美國(guó)韋恩州立大學(xué)Arava等[31]是第一批關(guān)注貴金屬催化劑鉑(Pt)對(duì)LiPSs氧化還原反應(yīng)的催化活性的,他們將Pt催化劑納米顆粒均勻分散在石墨烯層上,如圖3(a)所示。含硫的Pt/石墨烯復(fù)合物電極在初始放電過(guò)程中的容量為1100mA·h/g,在100次循環(huán)后保持在789mA·h/g;優(yōu)于初始放電比容量為740mA·h/g、在100 次循環(huán)后為580mA·h/g 的作為對(duì)照的石墨烯電極。為了考察Pt 的催化作用,從Tafel圖中計(jì)算出交換電流密度(io),如圖3(b)所示,Pt/石墨烯電極放電時(shí)io為3.18mA/cm2,石墨烯電極放電時(shí)io僅為1.18mA/cm2。在逆反應(yīng)中,Pt/石墨烯電極的io同樣高于石墨烯電極,說(shuō)明Pt可在充放電過(guò)程中促進(jìn)LiPSs的氧化還原反應(yīng)。
金屬納米催化劑通過(guò)氧化還原過(guò)程促進(jìn)了不溶態(tài)Li2S2/Li2S 向長(zhǎng)鏈LiPSs 的轉(zhuǎn)化,阻止了Li2S2/Li2S在電極上的沉積,促進(jìn)了反應(yīng)動(dòng)力學(xué)。孫世剛課題組[32]發(fā)現(xiàn),與純石墨烯相比,Co@GC-PC的存在促進(jìn)了放電過(guò)程中LiPSs的吸附,作用機(jī)理如圖3(c)所示。密度泛函理論計(jì)算表明,Co 與Li2S 之間存在較強(qiáng)的相互作用,使得Li2S 在Co(111)上的分解勢(shì)壘較低,有利于其在放電過(guò)程中的沉積和充電過(guò)程中的氧化。
另外,萬(wàn)立駿課題組[33]發(fā)現(xiàn)單分散Co 原子嵌入N摻雜石墨烯(Co-N/G)可以觸發(fā)LiPSs的表面介導(dǎo)反應(yīng),使得S@Co-N/G電極在具有90%的高硫質(zhì)量分?jǐn)?shù)時(shí),仍可以提供1210mA·h/g 的比容量以及硫載量高達(dá)6.0mg/cm2時(shí),仍然具有每循環(huán)0.029%的低衰減率。圖3(d)利用操作X射線吸收光譜和第一性原理計(jì)算相結(jié)合,證明了Co-N-C配位中心作為雙功能電催化劑,分別促進(jìn)放電和充電過(guò)程中Li2S的形成和分解過(guò)程。
張躍鋼課題組[34]以原子分散的單個(gè)Fe 原子為催化劑,促進(jìn)了相對(duì)惰性的Li2S陰極在長(zhǎng)期循環(huán)過(guò)程中的脫鋰過(guò)程,加速了可逆的電化學(xué)轉(zhuǎn)化反應(yīng)。12C下容量可達(dá)到588mA·h/g,5C下1000次循環(huán)的平均容量衰減率為0.06%,證明了單原子催化在提高鋰硫電池性能方面的有效性。
2.5 碳化物與磷化物
碳化物中的過(guò)渡金屬原子對(duì)LiPSs 的轉(zhuǎn)化具有很高的催化活性,其表面具有較強(qiáng)的親硫性,因此經(jīng)常作為錨定材料來(lái)有效地吸附LiPSs。金屬磷化物被認(rèn)為是鋰硫電池中最有前途的催化劑之一,由于親脂相互作用形成P—Li鍵,促進(jìn)硫代硫酸鹽的形成,有利于短鏈Li2S2/Li2S的生成[35]。
Zhang 等[36]將Fe3C 納米粒子嵌入N 摻雜多孔碳片中(Fe3C@NPCS),由于Fe3C 納米粒子具有較好的電導(dǎo)率,可以通過(guò)快速電子傳遞加速LiPSs 的電化學(xué)反應(yīng),作用機(jī)理見(jiàn)圖4(a)。另外,通過(guò)密度泛函理論計(jì)算證實(shí),F(xiàn)e3C納米粒子能夠通過(guò)強(qiáng)Fe—S化學(xué)鍵對(duì)LiPSs進(jìn)行強(qiáng)吸附。
Yang 等[37]證明了磷化物FeP 能與LiPSs 形成較強(qiáng)的化學(xué)鍵,該納米晶體對(duì)LiPSs 的轉(zhuǎn)化反應(yīng)和較低的Li2S成核能壘具有良好的催化作用。
此外還有B4C[38]、CoP[39]等材料均能表現(xiàn)出良好的性能和催化效果。
2.6 金屬有機(jī)框架與共價(jià)有機(jī)框架
金屬有機(jī)框架(MOFs)是由有機(jī)配體和金屬離子或團(tuán)簇通過(guò)配位鍵自組裝形成的具有分子內(nèi)孔隙的有機(jī)-無(wú)機(jī)雜化材料。具有較高的比表面積、不飽和的金屬位點(diǎn)和良好的孔隙。共價(jià)有機(jī)框架(COFs)是一種新興的多孔結(jié)晶框架,由輕元素(C、B、O 和N)組成,以共價(jià)鍵連接,具有規(guī)律的結(jié)構(gòu)周期性、固有的孔隙率、結(jié)構(gòu)的高度可調(diào)性和可設(shè)計(jì)性[40]。由強(qiáng)鍵構(gòu)成的高度連接的晶體結(jié)構(gòu)結(jié)合孔隙結(jié)構(gòu),使得構(gòu)建單元為網(wǎng)狀的MOFs 和COFs 的分子可以選擇容納高密度的固定陰離子,從而使鋰離子成為唯一的可移動(dòng)物種[41]。
董全峰課題組[42]合成了一種如圖4(b)所示的新型的含Co 和N 摻雜石墨碳(Co-N-GC)的金屬有機(jī)骨架衍生硫基體,具有大的比表面積,能夠同時(shí)催化硫的氧化還原和LiPSs 的捕獲,顯著提高了鋰硫電池的比容量、倍率性能和循環(huán)穩(wěn)定性。

圖4 Fe3C@NPCS復(fù)合陰極在充放電過(guò)程中的作用(a)[36]、雙催化劑錨定Co-N-GC復(fù)合材料的制備及其與LiPSs的相互作用(b)[42]、PTPPCo/MWCNT修飾層對(duì)LiPSs協(xié)同抑制和催化的多功能機(jī)理(c)[47]以及DCBQ在Li-S氧化還原反應(yīng)中的作用(d)[48]
Wang 等[43]提出了一種基于MOF(ZIF-67)衍生的原位摻氮碳納米籠(CoP-HNC)作為高效硫載體。空心極性雜原子N摻雜碳結(jié)構(gòu)具有豐富的孔隙和腔體,可有效緩解體積膨脹、緩沖電解質(zhì)和捕獲LiPSs。更重要的是,嵌套的極性CoP 納米顆粒作為電催化劑,不僅錨定了LiPSs 中間體,而且能夠顯著促進(jìn)LiPSs 轉(zhuǎn)化的氧化還原動(dòng)力學(xué)。CoPHNC 電極在5C時(shí)能提供約為800mA·h/g 的容量,1000 次循環(huán)后容量衰減率為0.02%,循環(huán)穩(wěn)定性提高。
張永光課題組[44]以MIL-88B為例,提出將無(wú)定型金屬有機(jī)骨架(aMOF)用于構(gòu)建鋰硫電池分離器。由于欠配位效應(yīng),aMIL-88B對(duì)硫類物質(zhì)具有較高的吸附能力和催化活性,由aMIL-88B修飾的分離器實(shí)現(xiàn)高效和可逆的硫電化學(xué),表現(xiàn)出極好的循環(huán)性,1C下循環(huán)500圈后仍有740mA·h/g的高容量保留。采用自模板配位-復(fù)制的方法開(kāi)發(fā)了一種新型的三維有序宏觀微孔金屬有機(jī)骨架(3DOM ZIF-8)[45],作為提高鋰硫電池性能的高級(jí)硫儲(chǔ)層。獨(dú)特的分層結(jié)構(gòu)不僅有利于電解液的滲透和質(zhì)子的傳遞,而且增加了活性界面大量暴露的表面積。此外,納米ZIF-8 亞基與LiPSs 化學(xué)相互作用后對(duì)硫具有很強(qiáng)的固載和催化作用,從而能夠明顯抑制穿梭效應(yīng),并提高反應(yīng)動(dòng)力學(xué)。得益于這些協(xié)同特性,基于3DOM ZIF-8的硫電極表現(xiàn)出優(yōu)異的電化學(xué)性能,即循環(huán)穩(wěn)定性較長(zhǎng),500次循環(huán)后容量衰減率為0.028%,在提高硫負(fù)載和限制電解液的條件下,具有較高的面積容量和良好的循環(huán)性能。
張強(qiáng)課題組[46]采用以聚苯乙烯(PS)為模板制備了卟啉有機(jī)骨架空心球(POF-HSs)。POF-HSs得益于其極性化學(xué)結(jié)構(gòu)和空心球形形貌,通過(guò)化學(xué)吸附和物理約束的雙重作用,充分緩解了LiPSs 的穿梭現(xiàn)象,成為了硫陰極的理想主體材料,賦予鋰硫電池高容量和循環(huán)壽命長(zhǎng)的優(yōu)良性能。
張嘉恒等[47]通過(guò)真空過(guò)濾的方法將聚四苯基卟啉鈷(PTPPCo)吸附在多壁碳納米管(MWCNT)上,制備了功能性隔膜(PTPPCo/MWCNT/PP),MWCNT具有優(yōu)異的導(dǎo)電性,并可以促進(jìn)沉積在陰極上惰性硫的再利用。PTPPCo不僅為捕獲LiPSs提供了豐富有力的化學(xué)吸附位點(diǎn),而且在LiPSs 轉(zhuǎn)化反應(yīng)中發(fā)揮了優(yōu)異的催化作用,機(jī)理如圖4(c)所示。此外,PTPPCo/MWCNT 復(fù)合材料易被電解質(zhì)滲透,有利于鋰離子的快速擴(kuò)散。結(jié)合MWCNT和PTPPCo 的優(yōu)點(diǎn),具有PTPPCo/MWCNT/PP 的鋰硫電池可以通過(guò)物理約束和化學(xué)吸附的雙重作用有效地捕獲LiPSs,提高氧化還原轉(zhuǎn)化動(dòng)力學(xué),從而實(shí)現(xiàn)高硫利用率和轉(zhuǎn)化率、優(yōu)異的倍率性能,且循環(huán)性能穩(wěn)定持久,抗自放電能力增強(qiáng)。
2.7 其他有機(jī)分子
吉林大學(xué)李峰課題組[48]采用原位凝固策略,通過(guò)2,5-二氯-1,4-苯醌(DCBQ)在電解質(zhì)中引發(fā)親核取代反應(yīng)來(lái)有效地阻塞LiPSs。LiPSs 可以被DCBQ以固體有機(jī)硫的形式共價(jià)固定,使其有效地保留在陰極內(nèi),有助于提高容量保留率。此外,DCBQ中的苯并醌基能夠加速鋰離子的傳遞,促進(jìn)硫的氧化還原反應(yīng)動(dòng)力學(xué),其機(jī)理見(jiàn)圖4(d)。
Goodenough 團(tuán)隊(duì)[49]利用雙(4-硝基苯基)碳酸酯作為有機(jī)液體電解質(zhì)的添加劑與可溶性LiPSs 反應(yīng)生成不溶的Li2S2/Li2S 與4-硝基酚鋰;4-硝基酚鋰與鋰金屬陽(yáng)極反應(yīng),生成一個(gè)有益于鋰離子傳導(dǎo)的陽(yáng)極鈍化層。
Wei 等[50]將嗎啉分子成功接枝到商用聚丙烯分離器上。密度泛函理論(DFT)計(jì)算結(jié)果表明,嗎啉側(cè)鏈能均勻、可逆地吸附所有高階聚硫化合物。這種雙原子化學(xué)吸附調(diào)節(jié)了硫相關(guān)化合物之間的轉(zhuǎn)化,加速了Li2S的成核過(guò)程。改進(jìn)后的隔膜電池在0.5C下循環(huán)500次后,放電容量高達(dá)827.8mA·h/g,如圖5所示。
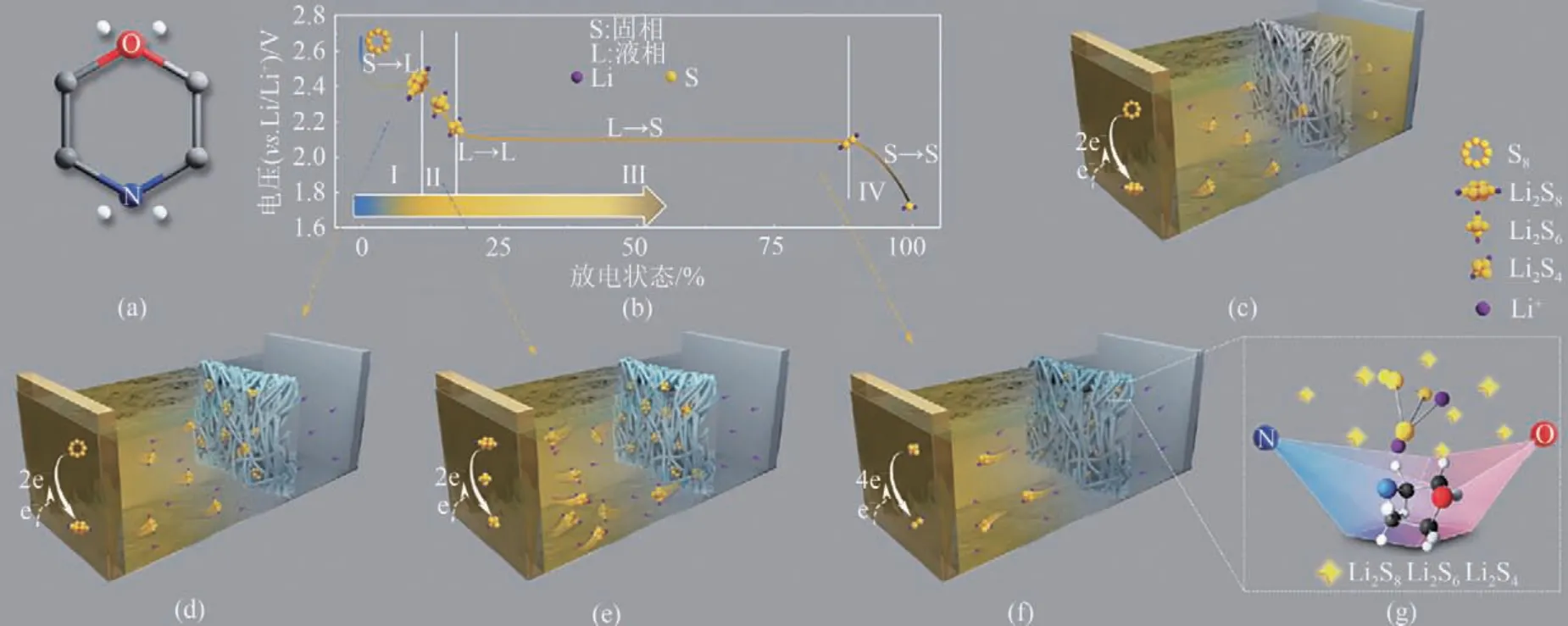
圖5 嗎啉的化學(xué)結(jié)構(gòu)(a)、LiPSs在放電過(guò)程中對(duì)電勢(shì)的演化(b)、PP分離器不能阻止溶解的LiPSs從陽(yáng)極側(cè)擴(kuò)散的演示(c)、PP-C-St-MP分離器固定溶解的LiPSs并釋放需要從第Ⅰ階段到第III階段在陰極側(cè)轉(zhuǎn)換時(shí)的LiPSs(d)~(f)以及嗎啉的雙原子化學(xué)吸附所實(shí)現(xiàn)的可逆吸附效應(yīng)(g)[50]
另外,Wei 等[51]以“軟硬酸堿理論”為基礎(chǔ),在商業(yè)隔膜上將三級(jí)胺覆蓋層(TAL)與LiPSs選擇性配位。用于捕獲LiPSs 的錨定基團(tuán)TAL 化學(xué)接枝到隔膜上,改變LiPSs 反應(yīng)路徑的同時(shí)又能保證有足夠的孔,不會(huì)阻礙鋰離子的傳輸,如圖6 所示。因正電荷主要分散在團(tuán)聚的LiPSs 分子的表面上,所以LiPSs是相對(duì)“軟”的酸。當(dāng)LiPSs進(jìn)一步還原并釋放到正極時(shí),在隔膜上構(gòu)建的“軟”基TAL可以錨定LiPSs 軟酸。而鋰離子的尺寸小、電荷密度高,通常被認(rèn)為是“硬”酸,它將被隔膜上的“軟”基TAL 排斥。靈活的“多硫化物鉗”構(gòu)造方法為實(shí)現(xiàn)高能量密度鋰硫電池鋪就了一條新途徑。
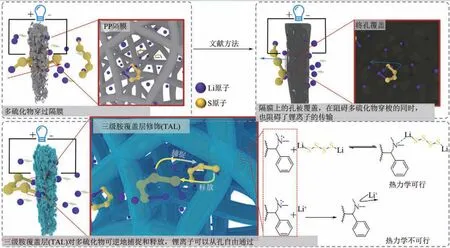
圖6 鋰硫電池隔膜的改性研究以及對(duì)LiPSs穿梭效應(yīng)抑制機(jī)理[51]
2.8 導(dǎo)電性載體
張強(qiáng)課題組[52]通過(guò)熱解生長(zhǎng)在石墨烯上的二維卟啉有機(jī)框架(POF),制備了N 摻雜碳納米片(PNG)用作中間層。由于POF的二維形貌,N元素完全暴露在石墨烯表面,而不受石墨烯骨架內(nèi)電子傳遞的阻礙。原子表面被完全暴露的親鋰位點(diǎn)修飾,以提供對(duì)LiPSs的強(qiáng)化學(xué)吸附,并改善電解質(zhì)的潤(rùn)濕性,而石墨烯襯底提供快速的電子傳遞,以促進(jìn)硫物種的氧化還原動(dòng)力學(xué),如圖7(a)所示。另外,他們還提出在還原氧化石墨烯納米片(CoSe2/G)上原位生長(zhǎng)鈣礬石CoSe2納米點(diǎn),構(gòu)建獨(dú)特的電解質(zhì)/CoSe2/G 三相界面[53]。該三相界面能夠提供強(qiáng)的化學(xué)吸附、高的電導(dǎo)率和極好的電催化LiPSs 氧化還原反應(yīng)。有效地增強(qiáng)了可溶LiPSs 的動(dòng)力學(xué)行為,調(diào)節(jié)了大電流密度下固體Li2S沉淀的均勻形核和可控生長(zhǎng),如圖7(b)所示。
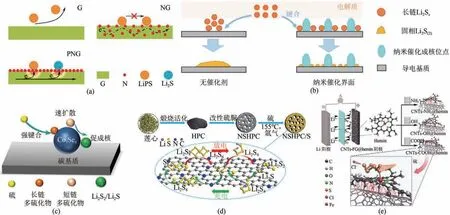
圖7 PNG對(duì)LiPSs的作用(a)[52]、在常規(guī)導(dǎo)電表面和具有均勻形核位點(diǎn)的導(dǎo)電和催化納米三相界面上的Li2S成核和生長(zhǎng)(b)[53]、LiPSs在N-CN-750@Co3Se4-0.1mol·L-1基體上的吸附和轉(zhuǎn)化(c)[59]、N,S共摻雜層次多孔碳/硫(NSHPC/S)復(fù)合材料的制備及與LiPSs相互作用(d)[61]、基于三種CNTs-FG@hemin陰極(FG=NH2,OH,COOH)的鋰硫電池結(jié)構(gòu)以及CNTs-COOH@hemin陰極上與LiPSs的作用機(jī)理(e)[63]
陳忠偉課題組[54]提供了一種利用天然豐富的有機(jī)分子蒽醌(AQ)抑制循環(huán)過(guò)程中氧化還原反應(yīng)中LiPSs的溶解和擴(kuò)散的策略。AQ很容易通過(guò)π-π相互作用與還原的氧化石墨烯(rGO)相連,隨后與硫(S-AQ-G)混合。AQ 和LiPS 的酮基之間的強(qiáng)Lewis 堿-酸相互作用改善了氧化還原動(dòng)力學(xué),減少了極化。因此,S-AQ-G電極比硫與還原氧化石墨烯(S-G)復(fù)合電極具有更高的速率能力和循環(huán)穩(wěn)定性。
3 催化性能提升策略
鋰硫電池的實(shí)際應(yīng)用還面臨著諸多挑戰(zhàn)[55]。單一策略很難達(dá)到鋰硫電池長(zhǎng)循環(huán)、低衰減的狀態(tài),為進(jìn)一步催化轉(zhuǎn)化多硫物種,采用雙重策略如具有電荷效應(yīng)(通過(guò)摻雜改變電子云密度)、吸附-催化共同作用(異質(zhì)結(jié))、雙相催化作用(分解和沉積)等電催化材料已經(jīng)被報(bào)道,它們不僅對(duì)LiPSs具有很強(qiáng)的親和力,還可以極大地增強(qiáng)電化學(xué)反應(yīng)動(dòng)力學(xué)。
3.1 摻雜與功能化
長(zhǎng)鏈可溶性LiPSs 和短鏈不溶的Li2S2/Li2S 末端硫均帶有負(fù)電荷而具有相同的極性性質(zhì)[56]。根據(jù)以往的研究發(fā)現(xiàn),雜原子摻雜可以改變碳基體的表面電子結(jié)構(gòu),使非極化碳基體材料變?yōu)闃O化碳基體材料[57]。這些極性材料可以提供足夠的電導(dǎo)率從而保證高硫利用率[35],其極性表面可以提供較強(qiáng)的親和力。因而,摻雜被認(rèn)為是一種有效的緩解穿梭效應(yīng)的方法。其中,N摻雜碳材料是最常用的單摻雜碳材料。
張強(qiáng)課題組[58]分析了LiPSs與N、O、P、S、B、F 和Cl 等各種單摻雜原子之間的偶極-偶極靜電相互作用,發(fā)現(xiàn)N或O摻雜的碳基體表現(xiàn)出增強(qiáng)的相互作用,可以阻止LiPSs 的來(lái)回移動(dòng)。Han 等[59]提出了將高導(dǎo)電性的Co3Se4納米粒子接枝到N摻雜3D碳基體表面制備的多功能硫基體,以抑制LiPSs 的穿梭,提高硫的利用率。通過(guò)調(diào)節(jié)碳基體和Co3Se4的分布,N-CN-750@Co3Se4-0.1mol/L 具有豐富的極性位點(diǎn),對(duì)于LiPSs 吸收和硫的轉(zhuǎn)化促進(jìn)具有很好的表現(xiàn),其機(jī)理如圖7(c)所示。
除碳基單原子摻雜外,還經(jīng)常采用雙摻雜。對(duì)于雙摻雜,兩種雜原子在改善LiPSs 的電化學(xué)反應(yīng)中起著至關(guān)重要的作用。大多數(shù)雙摻雜碳材料都含有N 和B、P、S 或O。余桂華課題組[60]將植酸摻雜聚苯胺水凝膠設(shè)計(jì)合成了氮磷共摻雜碳(N,P-C)框架,作為無(wú)黏結(jié)劑的Li2S/N,P-C 陰極。N 和P共摻雜碳的三維多孔結(jié)構(gòu)為鋰離子的輸運(yùn)提供了連續(xù)的電子通道和分層的多孔通道。磷的摻雜還可以通過(guò)硫與碳骨架的強(qiáng)相互作用抑制穿梭效應(yīng),從而獲得較高的庫(kù)侖效率。與此同時(shí),P摻雜在碳骨架中對(duì)改善反應(yīng)動(dòng)力學(xué)起著重要作用,它可以催化硫物種的氧化還原反應(yīng),降低電化學(xué)極化,提高Li2S的離子電導(dǎo)率。
王先友課題組[61]通過(guò)熱解法和水熱法成功合成了N、S 共摻雜分層多孔生物質(zhì)碳(NSHPC)的蜂窩狀結(jié)構(gòu),如圖7(d)所示,這種雙摻雜載體為溶解的LiPSs提供了化學(xué)吸附和活性位點(diǎn)。此外,N、S雙摻雜還提高了多孔碳的親水性和電導(dǎo)率,最終顯著提高了鋰硫電池的硫利用率和長(zhǎng)期循環(huán)性能。
此外,Liu 等[62]研究了一系列金屬/N 摻雜的半石墨有序介孔碳(Me-N-GOMCs,Me=Fe、Co、Ni和Cu)被設(shè)計(jì)為具有豐富孔隙率和高導(dǎo)電性的硫基質(zhì)。結(jié)果表明,F(xiàn)e 和N 的摻入可以顯著地增強(qiáng)LiPSs的固碳性能。此外,將Fe-N-GOMC/S復(fù)合材料植入碳紙(CPs)的空隙空間內(nèi),構(gòu)建了具有連續(xù)導(dǎo)電三維網(wǎng)絡(luò)結(jié)構(gòu)的獨(dú)立集成硫陰極。快速的離子/電子傳遞和氧化還原動(dòng)力學(xué)使鋰硫電池在高負(fù)載下具有良好的硫利用率。結(jié)果表明,在0.5C下,CP/Fe-N-GOMC/S電極的初始容量高達(dá)1473mA·h/g,且在500次循環(huán)后容量衰減率可達(dá)0.075%。
楊植課題組[63]將血晶素(hemin)分子接枝到用官能團(tuán)改性的碳納米管(CNTs-COOH、CNTs-OH 和CNTs-NH2)上,合成了新型仿生分子催化劑并應(yīng)用于鋰硫電池正極材料中。其中,CNTs-COOH 與hemin 形成p-p 共軛和配位鍵,能提高硫的導(dǎo)電性和分散性,加之通過(guò)Fe—O 鍵配位的Fe(III)絡(luò)合物對(duì)LiPSs 具有很高的吸附能力,可促進(jìn)LiPSs 的快速轉(zhuǎn)化,并能有效抑制穿梭效應(yīng),見(jiàn)圖7(e)。
蘇寶連課題組[64]通過(guò)親核攻擊與含氧官能團(tuán)連接的幾種碳原子來(lái)修飾帶有乙二胺基團(tuán)的碳納米管網(wǎng)絡(luò)。交聯(lián)胺化碳納米管框架為活性材料的快速電荷傳遞提供了途徑,增強(qiáng)了活性材料的反應(yīng)動(dòng)力學(xué),大大提高了活性材料的反應(yīng)速率和硫的利用率。通過(guò)密度泛函理論計(jì)算證實(shí),乙二胺基團(tuán)的引入促進(jìn)了表面極性的增強(qiáng),從而有效地防止LiPSs溶解,提高循環(huán)穩(wěn)定性。外部聚苯胺層從結(jié)構(gòu)上抑制聚硫化物,以防止穿梭效應(yīng)和活性物質(zhì)損失。
3.2 異質(zhì)結(jié)
金屬碳化物、氮化物和磷化物是理想的介質(zhì)材料,具有優(yōu)越的導(dǎo)電性,有助于表面電荷傳輸和加速氧化還原反應(yīng)。同時(shí)具有適度的表面極性,以平衡表面結(jié)合和擴(kuò)散。但與LiPSs 的結(jié)合能相對(duì)較低,其吸附能力受到影響;而表面擴(kuò)散緩慢的金屬氧化物與LiPSs 具有較高的結(jié)合能。利用二者的結(jié)合構(gòu)建高吸附性異質(zhì)結(jié)構(gòu)的材料,可以有效地提高LiPSs 的整體吸附性。特別是由于MXene 具有奇特的二維特性、極好的金屬導(dǎo)電性和豐富的表面可調(diào)功能,是一種理想的異質(zhì)介質(zhì)[25]。
Nazar等[65]證明當(dāng)表面反應(yīng)性的—OH基團(tuán)被硫取代時(shí),MXene 能夠通過(guò)S—Ti—C鍵與硫鍵結(jié)合;Ti2C表面的電子轉(zhuǎn)移加速了表面氧化還原反應(yīng)。據(jù)此提出了兩步作用機(jī)制:端位—OH的MXene 相首先發(fā)生—OH 與LiPSs 的氧化還原反應(yīng),形成表面硫代硫酸鹽基團(tuán),通過(guò)Wackenroder 反應(yīng)進(jìn)一步暴露Ti原子,然后亞穩(wěn)Ti原子與額外的LiPSs通過(guò)路易斯酸堿相互作用反應(yīng)生成S—Ti—C/N 化學(xué)鍵,如圖8(a)所示。
楊全紅課題組[66]通過(guò)原位制備TiO2-MXene(Ti3C2Tx)異質(zhì)結(jié)構(gòu),提出了一種對(duì)LiPSs具有強(qiáng)吸附能力和優(yōu)異轉(zhuǎn)化活性,具有高比表面積和電子導(dǎo)電性等優(yōu)點(diǎn)的多功能催化劑,如圖8(b)所示。在MXene 表面均勻分布的TiO2作為俘獲中心固定LiPSs,其異質(zhì)界面保證了錨定的LiPSs從TiO2快速擴(kuò)散到MXene,并賦予了端氧的MXene 表面高催化LiPSs 轉(zhuǎn)化的活性。另外,他們還證明了氧化層的摻雜能夠提高氮化物的催化活性[67]。硫摻雜的氮化鈦(TiN)氧化層,形成的Ti—S 鍵能夠從導(dǎo)電的Ti—N 基體上傳遞電子,從而保證了高催化活性。Ti—S 與Ti—O 鍵在原子水平上的結(jié)合有助于同時(shí)實(shí)現(xiàn)LiPSs的強(qiáng)捕獲和快速轉(zhuǎn)換。
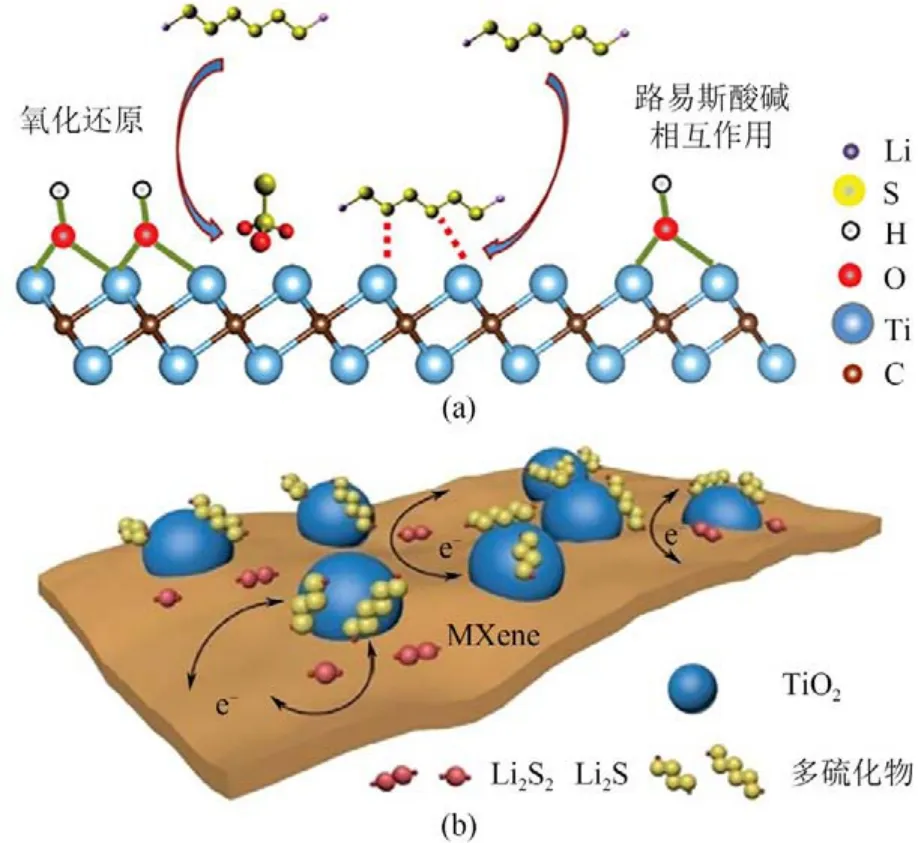
圖8 帶—OH的Mxene與LiPSs的相互作用(a)[65]以及TiO2-Ti3C2Tx結(jié)構(gòu)上的LiPSs捕獲和轉(zhuǎn)化(b)[66]
Cheng 等[68]將 石 墨 烯@Mo2C (GCF-G@Mo2C)異質(zhì)結(jié)構(gòu)的三維分層石墨泡沫碳作為鋰硫電池的獨(dú)立電極。N摻雜泡沫碳被具有類似木耳的多孔結(jié)構(gòu)的石墨烯片包裹,可以促進(jìn)電子和離子的快速傳遞,同時(shí)通過(guò)強(qiáng)極性相互作用的物理約束和化學(xué)吸附有效地錨定LiPSs。此外,如圖9 所示,Mo2C 納米粒子可以作為化學(xué)錨定中心提供強(qiáng)的LiPSs 吸附,其催化作用加速了LiPSs 轉(zhuǎn)化的氧化還原動(dòng)力學(xué),有助于提高速率性能。
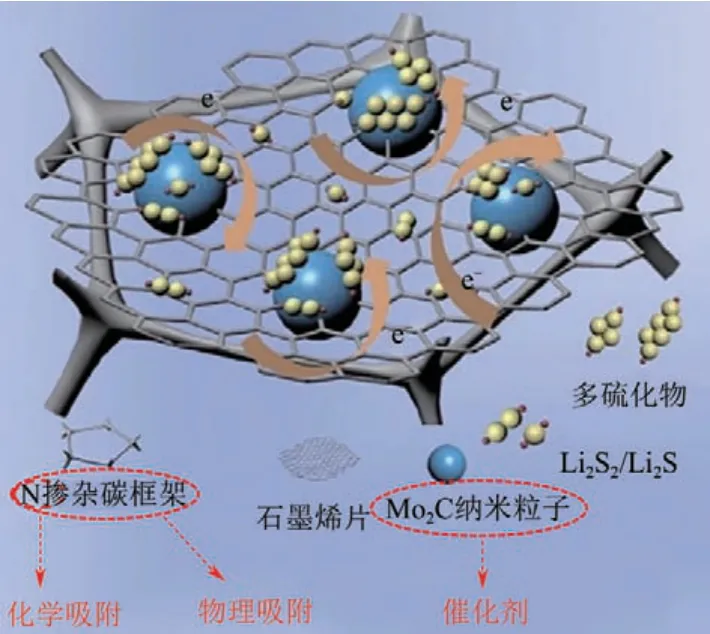
圖9 GCF-G@Mo2C電極的制備過(guò)程及與LiPSs作用機(jī)理示意圖[68]
吉林大學(xué)李峰課題組[69]將極性導(dǎo)電材料MoC@MoOx用于在氧化還原反應(yīng)中LiPSs 的固定化和轉(zhuǎn)化。MoOx的極性殼層能夠?qū)iPSs進(jìn)行化學(xué)吸附,MoC 的導(dǎo)電核有助于可溶性LiPSs 向Li2S 的轉(zhuǎn)化。將MoC@MoOx材料摻入碳纖維泡沫電極中,電化學(xué)性能顯著提高,具有較高的比容量和良好的循環(huán)穩(wěn)定性。
4 催化劑評(píng)價(jià)方法
4.1 循環(huán)伏安法
循環(huán)伏安法(CV)作為最基本的電化學(xué)研究方法之一,測(cè)試電池的極化電流與電極電勢(shì)之間的關(guān)系,可用于研究電極反應(yīng)的性質(zhì)、機(jī)理和反應(yīng)動(dòng)力學(xué)等。其原理是通過(guò)控制和改變電位以得到氧化還原電流方向。鋰硫電池中存在復(fù)雜的電化學(xué)反應(yīng),因而根據(jù)CV 曲線上的具體特征分析氧化還原過(guò)程的細(xì)節(jié),深入了解其中機(jī)理至關(guān)重要。
結(jié)合鋰硫電池中的充放電曲線,采用CV 曲線描述電化學(xué)氧化還原過(guò)程。圖10(a)為典型鋰硫電池中觀察到的CV曲線[70]。根據(jù)元素硫的反應(yīng)機(jī)理,2.4~2.2V 處的首個(gè)陰極峰對(duì)應(yīng)著S8到LiPSs 的液固轉(zhuǎn)化過(guò)程,在2.1~1.9V 的第二個(gè)陰極峰對(duì)應(yīng)著LiPSs還原成Li2S2和Li2S[71],這兩個(gè)還原過(guò)程對(duì)應(yīng)著放電過(guò)程中的兩個(gè)電壓平臺(tái)。在2.2~2.5V處的陽(yáng)級(jí)峰對(duì)應(yīng)于Li2S2/Li2S 氧化為L(zhǎng)iPSs/S 的過(guò)程[72],陽(yáng)級(jí)峰在這個(gè)范圍內(nèi)極有可能重合。

圖10 鋰硫電池中典型的CV曲線及NG/CNTs、NG/CNTs-COOH和NG/CNTs-S 電極的CV測(cè)試[70,73]
鋰離子遷移系數(shù)(DLi+)可用于比較氧化還原反應(yīng)動(dòng)力學(xué)。CV測(cè)試中,電流(i)與不同的掃描速率的關(guān)系符合式(4)。

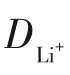
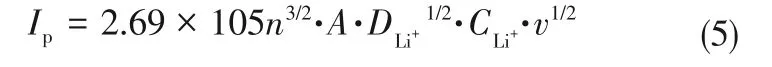


圖11 NG/CNTs、NG/CNTs-COOH和NG/CNTs-S電極在不同速率下的CV測(cè)試及Ip隨的變化曲線[73]
另外,可以采用對(duì)稱電池的CV 測(cè)試評(píng)價(jià)反應(yīng)動(dòng)力學(xué)。對(duì)稱電池由兩個(gè)不負(fù)載硫的完全相同的電極組成,多以Li2S6為電解液添加劑。開(kāi)路電壓(OCV)設(shè)置為0,從OCV 掃描一個(gè)對(duì)稱的電壓區(qū)間,如-0.8~0.8V 或-1.2~1.2V。通過(guò)比較電流密度的大小,即可評(píng)價(jià)氧化還原動(dòng)力學(xué)的快慢。
4.2 恒電流間歇滴定技術(shù)
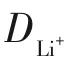
恒電流間歇滴定技術(shù)(GITT)[74]是一種暫態(tài)測(cè)量技術(shù),由德國(guó)化學(xué)家Weppner提出。該方法假設(shè)擴(kuò)散過(guò)程主要發(fā)生在固相材料的表層,具體來(lái)說(shuō),GITT 研究的是“物質(zhì)的擴(kuò)散過(guò)程與電荷轉(zhuǎn)移”的關(guān)系,主要由兩個(gè)部分組成。其中第一部分為小電流恒流脈沖放電,為了滿足擴(kuò)散過(guò)程僅發(fā)生在表層的假設(shè),恒流脈沖放電的時(shí)間t要比較短,需要滿足式(6)。第二部分為長(zhǎng)時(shí)間地靜置,以讓Li+在活性物質(zhì)內(nèi)部充分?jǐn)U散達(dá)到平衡狀態(tài)。GITT 測(cè)試由一系列“脈沖+恒電流+弛豫”組成。

式中,L為材料的特征長(zhǎng)度;D為材料的擴(kuò)散系數(shù)[75]。
除了測(cè)定離子的擴(kuò)散系數(shù),GITT 常用于對(duì)電極反應(yīng)中的微觀動(dòng)力學(xué)信息進(jìn)行測(cè)定,在電化學(xué)方法與原理的基礎(chǔ)上側(cè)重分析電極和樣品的化學(xué)反應(yīng)與極化,以此為依據(jù)對(duì)不同充放電深度的極化及其可能發(fā)生的化學(xué)反應(yīng)分段研究,逐步分析各階段的差異,從而找到各階段影響極化的關(guān)鍵因素。GITT最大的優(yōu)勢(shì)在于測(cè)量樣品的真實(shí)度較高。
4.3 原位XRD技術(shù)
原位表征技術(shù)能夠提供關(guān)于結(jié)構(gòu)演化、氧化還原機(jī)理、固-電解質(zhì)界面相(SEI)形成、副反應(yīng)和鋰離子輸運(yùn)的信息。原位XRD 通常用于監(jiān)測(cè)在循環(huán)過(guò)程或溫度變化過(guò)程中的材料的結(jié)構(gòu)變化[76]。
Walu?等[77]通過(guò)原位XRD 技術(shù)檢測(cè)鋰硫電池的充電和放電過(guò)程,如圖12(a)、(b)所示。發(fā)現(xiàn)最初存在于電極中的所有單質(zhì)硫都被還原為可溶的LiPSs。在兩個(gè)放電平臺(tái)之間的區(qū)域,活性物質(zhì)仍以可溶性形式存在于電解質(zhì)中且未檢測(cè)到峰。Li2S信號(hào)開(kāi)始出現(xiàn)于2θ=11.61°、13.41°、19.01°和22.31°,分別對(duì)應(yīng)于反射(111)、(200)、(220)和(311)面。Li2S 信號(hào)強(qiáng)度逐漸增大,直到在完全放電狀態(tài)下達(dá)到最大值。這些寬峰意味著晶體太小或結(jié)晶不良,但都很好地集中在Li2S化合物預(yù)期的位置上。在充電過(guò)程中,Li2S 的強(qiáng)度逐漸降低,直到完全消失,證明了Li2S 可逆氧化為可溶的LiPSs。然而,由于Li2S 的絕緣性和不溶性,該化合物在電極中一直存在到高電荷狀態(tài)(約74%)。在2.5V電荷平臺(tái)的末端[圖12(d)],非常明確的單質(zhì)硫峰再次出現(xiàn),它們的強(qiáng)度逐漸增加,直到電池充滿電。
原位XRD 譜圖中等高線圖[78]明確地顯示了硫依次還原為L(zhǎng)i2S8、Li2S6,然后是Li2S4,最后是Li2S相,見(jiàn)圖12(e)。此外,LiPSs 特別是長(zhǎng)鏈LiPSs 的峰在整個(gè)循環(huán)過(guò)程中從未完全消失,導(dǎo)致了比容量的一小部分損失。令人驚訝的是,與常規(guī)的鋰硫電池相比,未反應(yīng)的長(zhǎng)鏈LiPSs 并沒(méi)有穿梭到鋰陽(yáng)極,也沒(méi)有進(jìn)一步反應(yīng),而是被應(yīng)用的玻璃纖維分離器吸附。
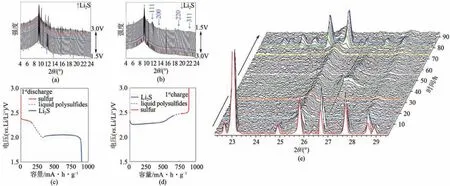
圖12 鋰硫電池在放電(a)和充電(b)過(guò)程中的XRD譜圖及放電(c)和充電(d)的電化學(xué)圖[77]以及鋰硫電池的原位XRD測(cè)試(e)[78]
4.4 自由基檢測(cè)
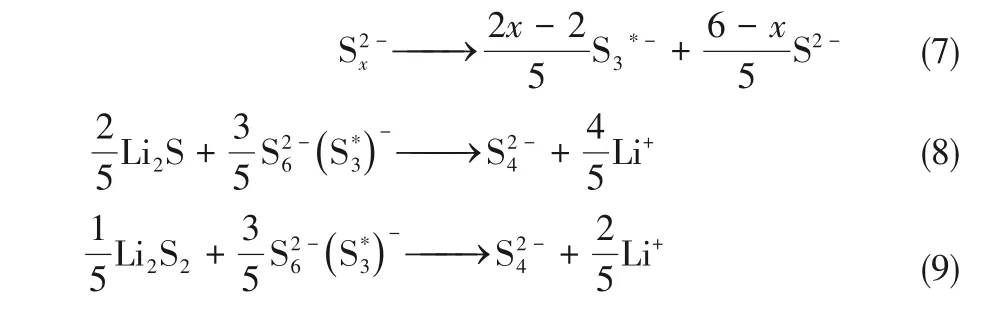
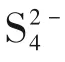
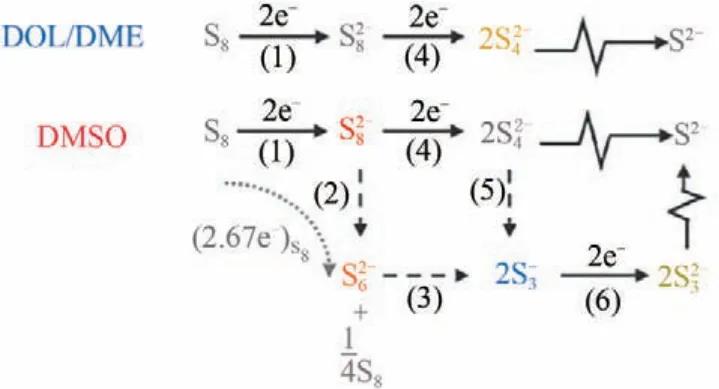
圖13 提出的依賴溶劑的硫還原反應(yīng)路徑[81]
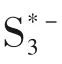
5 結(jié)語(yǔ)
對(duì)于鋰硫電池來(lái)說(shuō),可溶性LiPSs 與不溶的Li2S2/Li2S 之間的轉(zhuǎn)換緩慢是導(dǎo)致LiPSs 在電解質(zhì)中穿梭的主要原因,嚴(yán)重降低了電池的能量效率和循環(huán)性能。因此,理想的硫載體不僅需要有很強(qiáng)的親和力對(duì)LiPSs 進(jìn)行錨定,還需要有推動(dòng)LiPSs 向Li2S2/Li2S轉(zhuǎn)化、有效加速Li2S成核和分解的催化能力。為加速氧化還原動(dòng)力學(xué),促進(jìn)Li2S沉淀的均勻成核生長(zhǎng),多種催化材料已逐步應(yīng)用于鋰硫電池中。其催化機(jī)理可總結(jié)為以下幾個(gè)方面:①導(dǎo)電空間限制多硫化物的遷移,提升多硫化物利用效率;②增大催化材料的極性與活性位點(diǎn)的暴露,吸附多硫化物,加速成核過(guò)程;③化學(xué)鍵合作用錨定催化材料與多硫化物,改變多硫化物沉積-還原路徑;④提高電導(dǎo)率,加快電子傳遞,促進(jìn)活性物質(zhì)快速轉(zhuǎn)化;⑤生成硫自由基,利用均相化學(xué)反應(yīng)形成旁路反應(yīng)機(jī)理。
通過(guò)催化轉(zhuǎn)換這一策略抑制多硫化物的穿梭、促使多硫化物的快速轉(zhuǎn)化,是鋰硫電池未來(lái)的研究重點(diǎn)和電化學(xué)高密度儲(chǔ)能領(lǐng)域中的關(guān)注熱點(diǎn)之一,對(duì)鋰硫電池這一高效的儲(chǔ)能裝置發(fā)展具有重要意義,也將幫助推進(jìn)“碳達(dá)峰、碳中和”目標(biāo)的實(shí)現(xiàn)。
