UPLC-MS/MS測定人血漿中諾氟沙星的含量及其應用
2021-10-09 08:15:52劉海姣廖音娟唐智長沙市第四醫院長沙40006長沙都正生物科技有限責任公司長沙40000
中南藥學 2021年9期
關鍵詞:血漿
劉海姣,廖音娟,唐智(.長沙市第四醫院,長沙 40006;2.長沙都正生物科技有限責任公司,長沙 40000)
諾氟沙星是一種廣譜抗菌劑,已在多個國家使用,可有效治療人類和動物的感染,用于治療尿路感染、呼吸道感染、胃腸道感染、傷口感染等[1-5]。目前,有研究報道了海水、生物體液、食物以及人血漿中諾氟沙星的測定方法[4,6-8],常用的技術有紫外或熒光檢測的高效液相色譜法、毛細管電泳法、分光光度法、薄層色譜法、微生物法等[9]。Maia 等[10]使用基于HLB 柱的固相萃取(SPE)進行液相色譜-串聯質譜(LC-MS/MS)定量污水中7 種不同的氟喹諾酮類抗菌藥物,Ziarrusta 等[11]使用液相色譜串聯質譜法測定魚組織、生物體液和環境水中的氟喹諾酮類藥物。Helmy 等[12]用高效液相測定血漿中諾氟沙星的含量,Amjadi 等[13]使用硅摻雜碳點、鐵(Ⅱ)和K2S2O8組成的化學發光系統測定人血漿中的諾氟沙星,有研究采用LC-MS/MS 法并使用環丙沙星作為內標定量測定人血漿中諾氟沙星的濃度[14],也有報道采用超高效液相色譜-串聯質譜(UPLCMS/MS)法測定血漿中諾氟沙星的濃度[15],但所需分析時間普遍較長(4 min 以上),未完善對方法學的考察,如未進行樣品溶血基質效應、高血脂基質效應及全血穩定性的考察,這些因素均可影響到樣本測試結果的準確性和可靠性。因此建立一種準確、可靠的檢測人血漿樣品中諾氟沙星濃度的分析方法至關重要。
本試驗采用UPLC-MS/MS 法,以同位素為內標測定人血漿樣品中諾氟沙星的濃度,檢測過程更加簡單、快速,極大地縮減了分析時間和項目實施成本,可以更好地應用于諾氟沙星人體生物等效性研究。
1 材料
1.1 儀器
UPLC I-Class、Xevo TQD 質譜儀,配有電噴霧離子源,UNIFI 數據處理軟件(美國Waters 公司);MSA6.6S-CE 電子天平(Sartorius 公司);MilliQ Direct 8 超純水儀(Merck Millipore公司);ST16R 離心機(Thermo Fisher Thermo 公司);Pipetman NEO 和Pipet-Lite XLS+移液器(Gilson 和RAININ 公司);XW-80A 渦旋混合器(寧波新芝);Talboys 數顯多管式渦旋震蕩器(Troemner 公司);DTA-27 超聲波清洗器(鼎泰恒盛);GZX-9140MBE 電熱鼓風干燥箱(上海博迅);MLTS1368 醫用低溫冰箱(Thermo 公司)。
1.2 試藥
諾氟沙星(中國食品藥品檢定研究院,批號:13450-201206,純度:99.5%);內標諾氟沙星-d8(批號:12-ZCA-49-4,純度:97%,TRC)。甲醇、乙腈(色譜純,Merck 公司);甲酸(FA,色譜純,ACS);水為試驗室超純水儀自制水。
2 方法
2.1 色譜條件
色譜柱:Waters ACQUITY UPLC HSS T3(2.1 mm×50 mm,1.8 μm);流動相:0.2%甲酸水-乙腈=85∶15(V/V);流速:0.4 mL·min-1;運行時間:1.5 min;進樣量:10 μL;柱溫:40 ℃;自動進樣器溫度:20 ℃。
2.2 質譜條件
采用電噴霧離子源(ESI),正離子模式,MRM 監測,離子源溫度為150℃,毛細管電壓為0.5 kV,脫溶劑氣流為1000 L·h-1,溫度為500℃,諾氟沙星與內標諾氟沙星-d8 的離子對分別為m/z320.11 →m/z276.13 和m/z328.17 →m/z284.20。其中諾氟沙星的錐孔電壓為40 V,碰撞能為14 eV,駐留時間為0.08 s;諾氟沙星-d8 錐孔電壓為40 V,碰撞能為16 eV,駐留時間為0.08 s。
2.3 標準工作液及質控工作液的配制
精密稱取諾氟沙星對照品和諾氟沙星-d8 對照品適量(經折算),分別用50%甲醇水(含0.1%FA)溶解并定容至25 mL,搖勻,即得諾氟沙星儲備液和諾氟沙星-d8 儲備液。取諾氟沙星儲備液適量,用50%甲醇水(0.1%FA)稀釋,得含諾氟沙星質量濃度分別為16 000、8000、4000、2000、1000、800、400、200 ng·mL-1的系列濃度標準曲線工作液,12 800、6000、1600、600 ng·mL-1的系列濃度質控工作液及質量濃度為200 ng·mL-1的定量下限工作液。取內標諾氟沙星-d8 對照品儲備液,用50%甲醇水(含0.1%FA)稀釋,得質量濃度為10 μg·mL-1的內標溶液,取適量內標溶液用甲醇稀釋(內標溶液∶甲醇=3∶1000),得諾氟沙星-d8 質量濃度為30 ng·mL-1的內標工作液,同時也是蛋白沉淀劑。
2.4 標準曲線樣品及質控樣品配制
精密吸取人空白血漿190 μL,加入系列質量濃度工作液(16 000、8000、4000、2000、1000、800、400、200 ng·mL-1)或質控工作液 (12 800、6000、1600、600 ng·mL-1) 10 μL, 渦旋混勻,得到線性范圍濃度為10~800 ng·mL-1的標準曲線樣品以及濃度為30(LQC)、80(MQC)、300(HMQC)、640(HQC)ng·mL-1的質控樣品。
2.5 生物樣品處理方法
2.5.1 空白血漿樣品 精密吸取血漿樣品200 μL,加入甲醇400 μL,振蕩30 s,離心(18 400×g,8 min),取上清液100 μL,加稀釋劑(0.2%FA 水)400 μL,振蕩30 s 即得。
2.5.2 零濃度血漿樣品 精密吸取空白血漿200μL,加入400 μL 內標工作液,振蕩30 s,其余操作同“2.5.1”項下。
2.5.3 標準血漿樣品 精密吸取標準血漿樣品200μL,加入400 μL 內標工作液,振蕩30 s,其余操作同“2.5.1”項下。
2.6 方法學考察
2.6.1 專屬性 取空白血漿樣品、定量下限(LLOQ)濃度水平的樣品、只含內標的樣品以及只含待測物的定量上限(ULOQ)濃度水平的樣品。考察內源性物質及內標是否存在干擾。空白血漿中的內源性物質不對待測物與內標產生干擾;待測物與內標之間互相不產生干擾。結果見圖1。
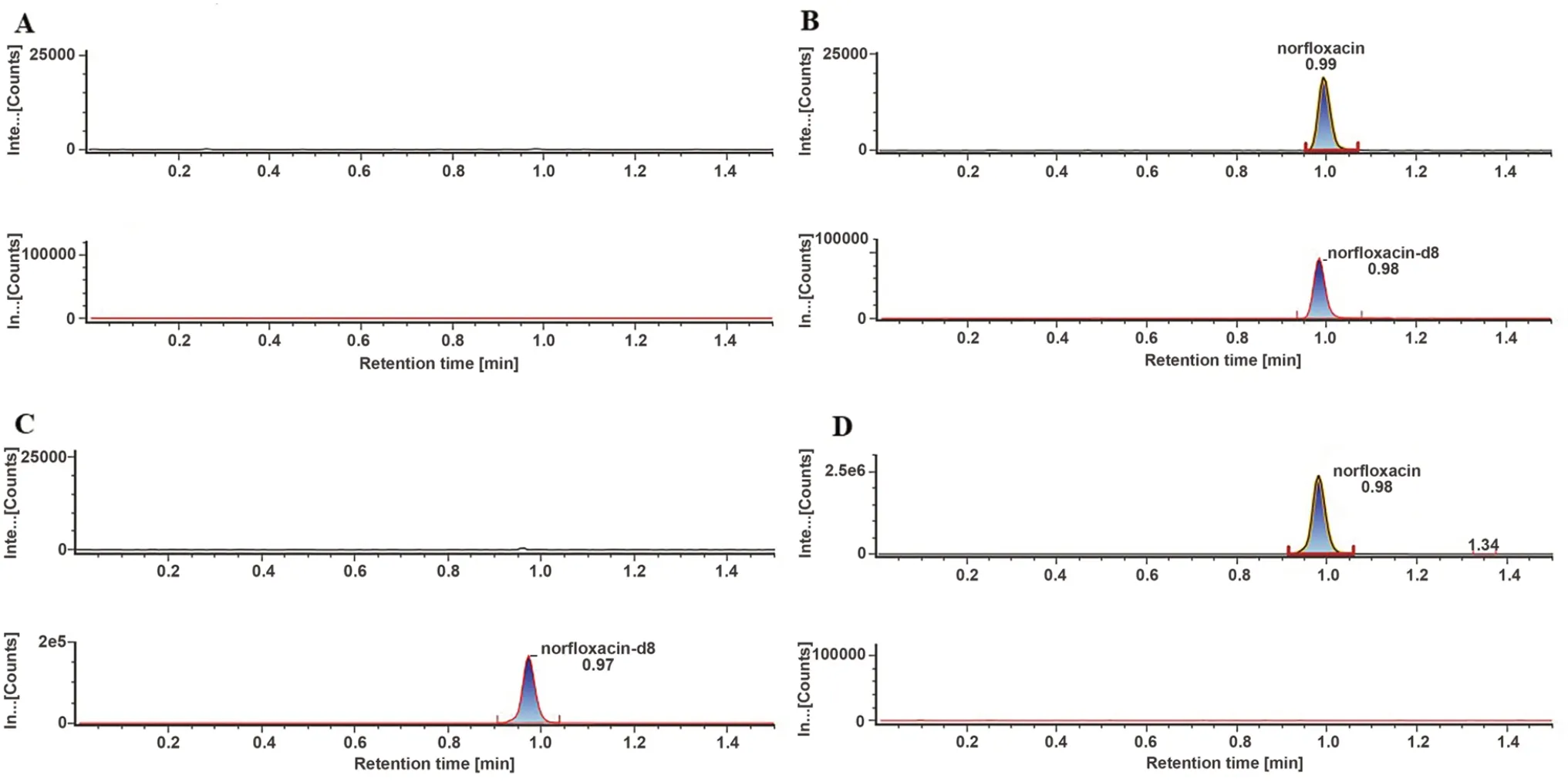
圖1 人血漿中諾氟沙星和諾氟沙星-d8 的典型色譜圖Fig 1 Typical chromatogram of norfloxacin and norfloxacin-d8 in human plasma
2.6.2 殘留考察 按定量上限樣品、空白樣品、定量下限樣品的順序進樣,記錄待測物與內標的色譜峰面積值,考察殘留情況。待測物進樣殘留小于20%且內標的進樣殘留小于5%。表明以本方法測試高濃度諾氟沙星人血漿樣品后,在系統中的殘留對于后續低濃度樣品的測試無影響。
2.6.3 線性范圍與定量下限考察 配制標準曲線血漿系列濃度樣品,按“2.5”項下方法處理,進樣分析,記錄待測物與內標的色譜峰峰面積值,以諾氟沙星與內標的色譜峰峰面積比值(Y)為縱坐標,以系列標準血漿樣品質量濃度(X)為橫坐標,進行線性回歸,得到回歸方程及相關系數。
結果諾氟沙星在10~800 ng·mL-1與測定值線性關系良好,定量下限為10 ng·mL-1,回歸方程為y=0.0227x-0.0037,r2=0.9995。
2.6.4 精密度與準確度 配制質量濃度為10、30、80、300、640 ng·mL-1的質控標準血漿樣品各6 份,按“2.5”項下方法處理后進樣測定,計算各樣品濃度,計算批內和批間的精密度,結果見表1。
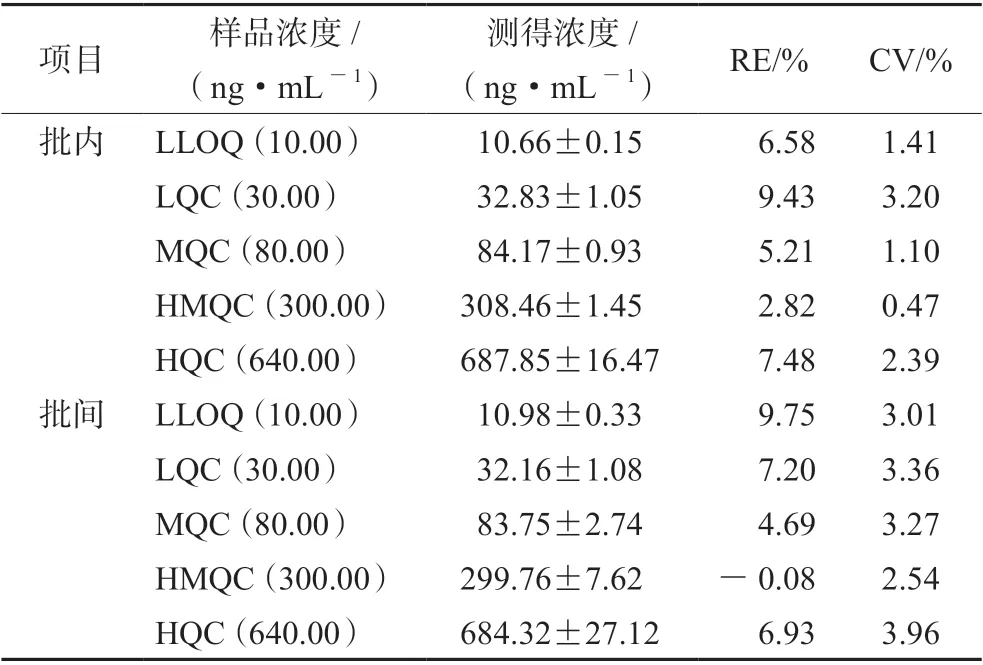
表1 人血漿樣本中諾氟沙星的批內、批間精密度(x±s,n =6)Tab 1 Intra- and inter-batch precision of norfloxacin in human plasma (x±s,n =6)
2.6.5 提取回收率 取6 份不同來源的空白血漿190 μL,每個來源平行2 份,加入甲醇400 μL,振蕩30 s,離心(18 400×g,8 min),得到6 份基質上清液,用0.2%FA 水配制含諾氟沙星低、中、高質量濃度溶液2.5、6.667、53.333(ng·mL-1)、和內標(5 ng·mL-1)。振蕩30 s,混勻,進樣10 μL,記錄待測物諾氟沙星與內標諾氟沙星-d8 的色譜峰峰面積值。
諾氟沙星質控樣品低、中、高濃度的回收率分別為82.7%,86.0%、82.7%,總體變異系數(CV)為2.30%。內標平均回收率為81.6%。
2.6.6 基質效應 取6 份不同來源的空白血漿190 μL,加入10 μL 50%甲醇水(0.1%FA)溶液,再加入甲醇400 μL,振蕩30 s,離心(18 400×g,8 min),得6 份不同來源空白基質上清液備用。按空白血漿方式同樣處理可得溶血基質上清、高脂基質上清。
移取空白基質上清100 μL,加入諾氟沙星低、中、高濃度溶液400 μL,振蕩30 s,混勻,即得基質效應考察樣本。按照空白血漿方式同樣處理可得溶血血漿、高脂血漿基質效應考察樣本。
取67%甲醇水溶液100 μL,加入諾氟沙星低、中、高濃度溶液400 μL,振蕩30 s,混勻,即得基質效應參比樣本。
人空白血漿、溶血血漿、高脂血漿均無明顯基質效應。結果見表2。
表2 人血漿中諾氟沙星基質效應(x±s,n =6)Tab 2 Matrix effect of norfloxacin in human plasma ( ±s,n =6)
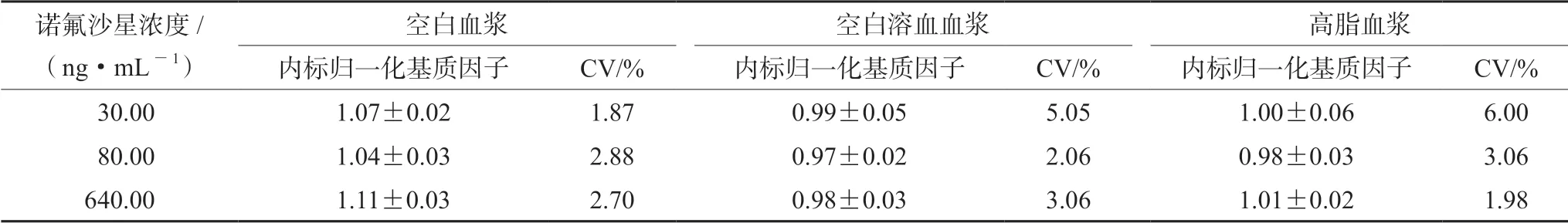
表2 人血漿中諾氟沙星基質效應(x±s,n =6)Tab 2 Matrix effect of norfloxacin in human plasma ( ±s,n =6)
諾氟沙星濃度/(ng·mL-1)空白血漿空白溶血血漿高脂血漿內標歸一化基質因子CV/%內標歸一化基質因子CV/%內標歸一化基質因子CV/%30.001.07±0.021.870.99±0.055.051.00±0.066.00 80.001.04±0.032.880.97±0.022.060.98±0.033.06 640.001.11±0.032.700.98±0.033.061.01±0.021.98
2.6.7 穩定性 對樣品采集、樣品儲存及分析過程中所涉及到的穩定性條件進行考察,結果見表3。
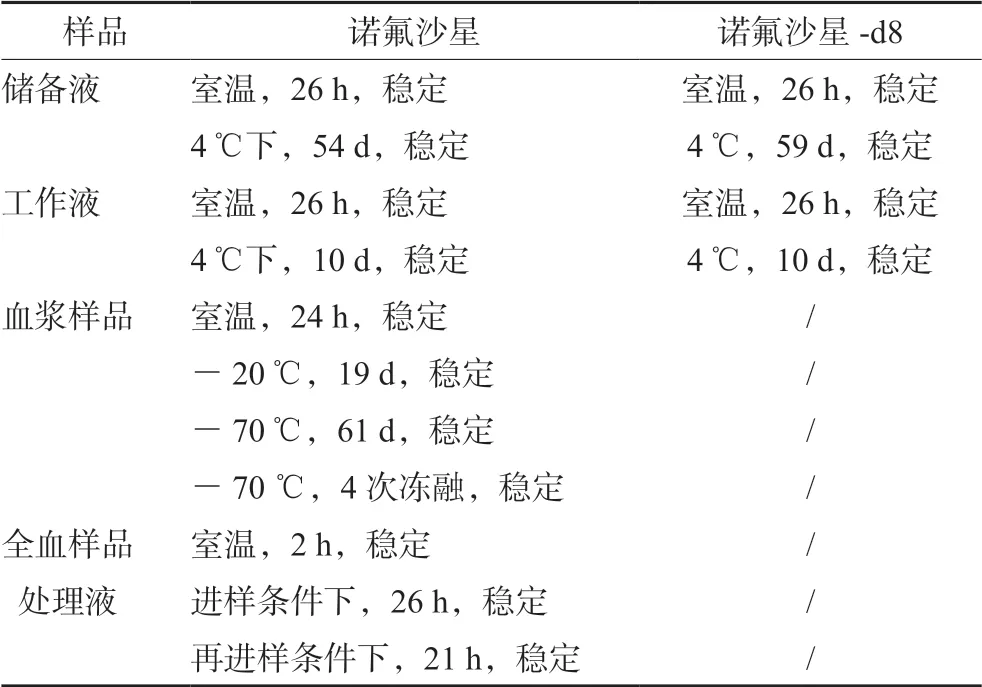
表3 諾氟沙星不同條件下的穩定性Tab 3 Stability test of norfloxacin under different conditons
2.6.8 稀釋方法學驗證 配制質量濃度為1600 ng·mL-1的諾氟沙星血漿樣品,用空白血漿稀釋10倍,平行處理6份,考察血漿樣品的稀釋可靠性。結果平均準確度偏差為-4.95%,精密度為2.1%。
2.6.9 分析批容量考察 配制標準曲線樣品及低、中、高濃度質控樣品,按照“2.5”項下血漿樣品處理方法處理后進樣分析,分析批進樣數量作為分析批容量考察結果。結果表明,分析批中可以連續進樣至少180 個未知樣本。
2.7 測定方法在生物等效性(BE)研究中的應用
2.7.1 給藥方案與血樣采集 本研究經湖南省腦科醫院醫學倫理委員會的批準(編號為2017010)。在一項單中心、隨機、開放、2×2 交叉設計的諾氟沙星BE 研究,入組8 例受試者且所有受試者均完成兩周期研究,洗脫期為7 d。受試者在早餐后單次口服受試制劑100 mg(1 片)或參比制劑100 mg(1 片),240 mL 常溫水送服。在給藥前(0 h)以及給藥后 0.25、0.5、0.75、1、1.25、1.5、2、3、4、5、6、8、10、12、24 h 進行血樣采集。血樣采集后置于已編號的肝素鈉抗凝負壓管內,輕柔顛倒混勻,4℃離心10 min(3500 r·min-1)后分離得血漿樣品,-80℃冰箱保存待測。
取受試者血漿樣本按“2.5.3”項下方法處理后進行測定,記錄待測物質和內標的峰面積,代入回歸方程,計算血藥濃度。
2.7.2 數據分析 血藥濃度數據采用Phoenix WinNonlin 軟件(Pharsight Corporation)進行非房室模型藥代動力學參數的估算分析,計算主要藥代動力學參數,以全面反映藥物在人體內吸收、分布、代謝和排泄的特點。
2.7.3 健康受試者的藥代動力學研究 8 名受試者單劑量口服諾氟沙星受試制劑及參比制劑后血漿中諾氟沙星平均藥濃度-時間曲線見圖2;主要藥代動力學參數見表4。AUC0~t和AUC0~∞指標均提示受試制劑與參比制劑生物等效。Cmax指標結果提示尚不能拒絕生物不等效,因本試驗設計密集采血點為0.25、0.5、0.75、1、1.25、1.5 h,實際結果受試制劑tmax中位值為2.25 h,Q1~Q3 為1.5~3 h;參比制劑tmax中位值為3 h,Q1~Q3 為2~3.5 h,密集采血時間早于受試制劑和參比制劑tmax、Cmax相關數據可能非真實值。本次為預試驗,樣本量較小,因此Cmax相關數據需要進一步驗證。
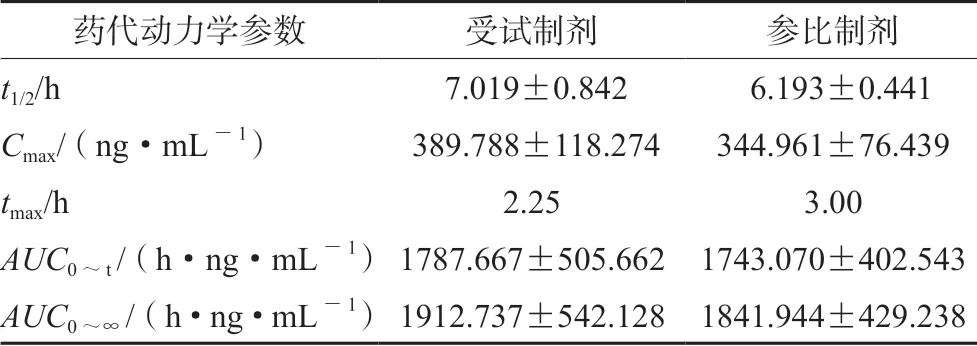
表4 8 名受試者單劑量口服諾氟沙星的主要藥代動力學參數(x±s)Tab 4 Main pharmacokinetic parameters of norfloxacin in 8 healthy volunteers after a single oral dose (x±s)
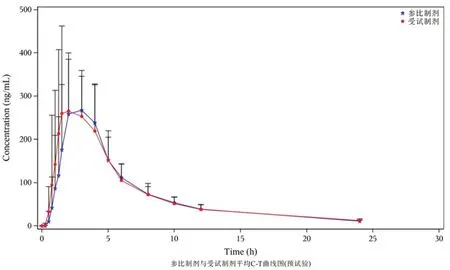
圖2 受試者口服受試制劑和參比制劑后血漿諾氟沙星平均血藥濃度-時間曲線(n =8)Fig 2 Mean plasma concentration-time curve of norfloxacin in the test and reference preparations after the oral administration (n =8)
3 討論
3.1 配制溶液及色譜柱的選擇
本方法的關鍵點是根據諾氟沙星的結構特點對其色譜、質譜條件進行優化。諾氟沙星在水中溶解度較小[16],《中國藥典》中也對其溶解性進行了闡述,根據這些信息,本方法選擇50%甲醇水(含0.1%FA)溶液配制諾氟沙星溶液。此外,諾氟沙星屬于喹啉羧酸類化合物,同時含有羧基和氨基,其logP為0.82,表明該化合物極性較大,在常規的C18柱中保留較差[17],所以在液相條件摸索過程中,選擇Waters ACQUITY UPLC HSS T3(2.1 mm×50 mm,1.8 μm),該色譜柱完全耐受100%水相且對含有羧酸基團的化合物有較好的保留。
3.2 流動相的選擇
本試驗過程中對流動相進行了優化,水相考察了不同比例甲酸水及緩沖鹽體系,通過對比,確定使用水相中含0.2%FA 最為適合。有機相分別用甲醇和乙腈進行洗脫,經過對比發現使用乙腈作為有機相時,色譜峰峰寬更窄,且響應更好。另外乙腈的洗脫能力比甲醇強,很好地避免了諾氟沙星在色譜柱中殘留。
3.3 質譜條件的考察
本試驗采用正離子模式,在流動相中加入適量甲酸,有助于提高離子化效率,提高響應,同時也能獲得較好的色譜峰,而負離子模式則響應較低,與峰形不能同時兼顧,且文獻也多采用正離子模式[14-15,18]。
3.4 內標的選擇
本試驗采用同位素內標,其被廣泛用于化學和臨床分析中的定量分析[19-20],同位素內標具有與分析物幾乎相同的物理化學性質,在分析物信號波動和量的損失上的矯正作用更強,可以有效校正甚至消除基質效應的影響。為保證待測物和內標之間不存在明顯干擾,本試驗對同位素內標和待測物之間的通道干擾進行了考察,最終選定m/z320.11 →m/z276.13 和m/z328.17 →m/z284.20 分別作為諾氟沙星和內標的檢測離子對。
3.5 分析效率的優化
本方法通過對色譜柱、流動相體系、前處理過程中稀釋劑的優化,避免了殘留。得益于樣品前處理過程的優化,使得本方法通過簡單的等度洗脫就可以實現很好的檢測結果,與采用梯度洗脫的方式相比,基線更平穩,樣品保留時間更穩定,也不容易出現殘留,而且對儀器的要求降低,即使單液相泵也可以正常運行該方法,另外樣品之間不需要設置平衡時間來讓系統恢復到所需的流動相比例,可以極大地縮短進樣時間,這對于樣品測定來說,相同時間內可以檢測更多的樣品,明顯提升測定效率,顯著降低儀器占用成本和人工成本。本方法將分析時間縮短為1.5 min,極大地提高了分析效率,非常適合于大批量樣品的分析。
3.6 小結
本次方法學驗證嚴格按照國內外法規要求進行,驗證內容完整且各項驗證內容均符合驗證接受標準。尤其是近年來備受關注的溶血基質效應、高脂基質效應和全血穩定性內容,本方法均進行了完整的考察,使得方法更加完善、可靠,且已用于諾氟沙星片人體BE 研究,樣品測試完成后還開展了試驗樣品再分析(ISR)測試,ISR 合格率為100%,表明該方法可以穩定、可靠、可準確地檢測人血漿樣品中諾氟沙星的濃度,為諾氟沙星BE 研究提供真實、可靠的血藥濃度數據。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
