次亞磷酸含量測定新方法的研究
2021-10-12 03:57:14周駿宏陳杭馨陳生悅萬宗靜
無機鹽工業 2021年10期
鄭 娜,周駿宏,陳杭馨,陳生悅,萬宗靜,盛 余
(黔南民族師范學院化學化工學院,貴州都勻558000)
磷在生產生活中的應用非常廣泛,對國民經濟有著極其重要的作用。磷含量作為磷化工行業的常規測定項目,其測定方法分為化學分析法和儀器分析法兩種。其中,儀器分析法中實驗室最常見的是分光光度法[1],主要又分為磷釩鉬黃法和磷鉬藍法及離子締合物法,在高端分析領域儀器分析法還有離子色譜法[2]和ICP-AES法[3-5];而化學分析方法則主要是喹鉬檸酮重量法和容量法,兩種都是實驗室常用的測磷含量的方法。其中重量法準確度極高,并且測量范圍廣(大約為30~130 mg/L),因而被定為仲裁法。
分光光度法測量樣品中磷含量,一般采用喹鉬檸酮法、磷釩鉬黃法、磷鉬藍法,都是基于形成磷鉬雜多酸的測定方法,其原理是正磷酸鹽在酸性條件下與鉬酸鹽等定量反應形成磷鉬雜多酸,通過測定磷鉬雜多酸的含量算出磷含量[6]。一般需要消解或灰化法處理樣品[7-8]。但在喹鉬檸酮重量法和容量法中,樣品的預處理卻鮮有研究,而其他容量法對次亞磷酸的測定一般采用的是氧化還原滴定法,但存在滴定終點不靈敏導致測定誤差較大的問題。而重量法測樣品的磷含量雖然具有準確性高的優點,但由于重量法目前少有預處理方法,所以只適用于正磷酸根的測定,對次亞磷酸完全不適用,所以不能直接采用重量法進行次亞磷酸的測定。
重量法測定磷組分含量具有準確度高的優點(尤其是對高含量樣品),但由于其存在不適用于低價磷的缺陷,即喹鉬檸酮重量法無法直接測量次亞磷酸及鹽的問題,筆者提出一種先用合適的氧化劑將次亞磷酸全部氧化為正磷酸,再用重量法測定的思路并進行探究。此法可適用于測定含磷量較高且含有以非正磷酸形式存在的磷樣品的總磷含量的準確測定,并且此新方法有準確度較高、適測范圍廣、可廣泛應用的特點。
1 實驗方法
1.1 主要儀器和試劑
儀器:分析天平、循環水多用真空泵、電熱恒溫鼓風干燥箱、G4玻璃坩堝、玻璃漏斗。
試劑:濃硝酸、0.1 mol/L硝酸鈰銨溶液、喹鉬檸酮沉淀劑。
喹鉬檸酮沉淀劑的配制:1)稱取70 g鉬酸鈉溶解于150 mL水中,此溶液為溶液A;2)稱取60 g檸檬酸溶解于150 mL水和85 mL硝酸的混合溶液中,此溶液為溶液B;3)在攪拌下將溶液A倒入溶液B中,此溶液為溶液C;4)在100 mL水中加入35 mL硝酸,再加入5 mL喹啉,此溶液為溶液D;5)將溶液D倒入溶液C中,混勻。放置12 h后,用玻璃砂坩堝過濾,再加入280 mL丙酮,用水稀釋至1 000 mL混勻,貯存于聚乙烯瓶中。
1.2 實驗步驟
1.2.1 氧化
取1.000 0 g(精確到0.000 1 g)試樣,置于100 mL小燒杯溶解,移入500 mL容量瓶,用水稀釋至刻度,搖勻,制得待測溶液。取待測溶液10 mL置于250 mL燒杯中,加入純水至100 mL,加入10 mL濃硝酸,再加入15 mL硝酸鈰銨溶液。在燒杯中放入玻璃珠或沸石,并蓋上表面皿,置于電熱板上加熱,使溶液沸騰15 min以上,確保溶液中次亞磷酸全部充分氧化,并且使過量的硝酸鈰銨分解。
1.2.2 沉淀
在氧化好的樣品溶液中加入5 mL濃硝酸,再加入50 mL喹鉬檸酮試劑。將其置于75℃水浴鍋中加熱數分鐘,停止加熱。取下燒杯,冷卻至室溫。用預先恒重好的玻璃坩堝抽濾,將沉淀轉移到玻璃坩堝中,置于250℃(±5℃)鼓風干燥箱中干燥至恒重,然后置于干燥器中冷卻20~30 min至室溫,稱量。
1.3 測定結果的計算
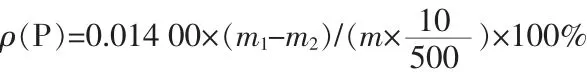
其中:0.014 00為磷鉬酸喹啉摩爾質量換算為磷摩爾質量的系數;m1為坩堝與沉淀的質量,g;m2為坩堝的質量,g;m為稱取的樣品質量,g。
2 結果與討論
2.1 磷鉬酸喹啉沉淀形成條件的探究
磷鉬酸喹啉是一種大分子的、溶解度很小的難溶鹽,該沉淀在硝酸酸性介質中生成,利用硝酸的氧化性來保證磷和沉淀劑中的鉬均以高價狀態存在。在酸性介質中,正磷酸根與喹鉬檸酮沉淀劑反應生成黃色磷鉬酸喹啉沉淀,經過濾、洗滌、干燥和稱量所得沉淀,根據沉淀質量換算出五氧化二磷的含量。喹鉬檸酮只能沉淀正磷酸,而對于待測樣品中的亞磷酸、次磷酸的成分,喹鉬檸酮無法測定。
為了驗證喹鉬檸酮與次磷酸、亞磷酸(根)等不同形態的磷是否正常響應而形成沉淀,分別用正磷酸、次磷酸、亞磷酸進行對照實驗,結果如表1所示。
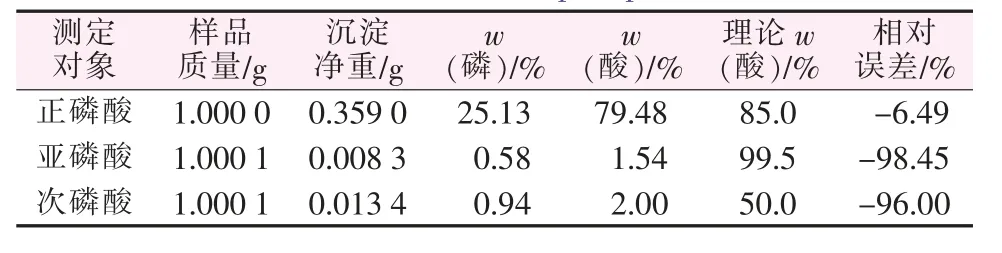
表1 原方法對不同形態的磷的測定Table 1 The original method for determination of different forms of phosphorus
可見,在次亞磷酸含量的測定中,原始的重量法根本無法得到沉淀,幾乎無法測出其含量,說明喹鉬檸酮不與次亞磷酸響應,原重量法不適用于測量次亞磷酸及鹽的磷含量,意味著采用傳統的重量法不可能測出次亞磷酸等低價態的磷的含量。這也是至今未看到有重量法運用在低價態磷測定上的報道的原因。沉淀反應方程式如下:
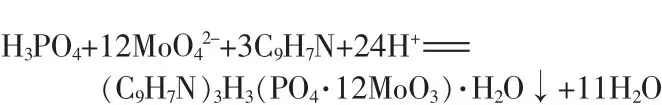
喹鉬檸酮沉淀劑僅能與五價磷作用,即使在實驗條件的強硝酸介質加熱情況下也一樣,而與三價、一價磷不反應,更不能形成沉淀。所以,要想采用喹鉬檸酮重量法測定低價形態的磷,只能先氧化低價磷將其完全轉化為五價磷,再進行測定。
2.2 氧化劑的篩選
本實驗在測定磷樣時,在使用沉淀劑喹鉬檸酮前使用氧化劑將亞磷酸、次磷酸全部氧化為正磷酸,再與沉淀劑喹鉬檸酮生成沉淀磷鉬酸喹啉(黃色)。發生的反應如下:
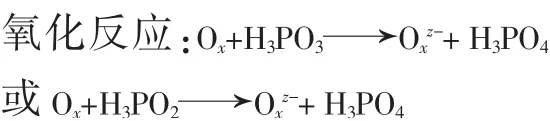
此時要準確測定磷含量就需選取合適的氧化劑,本實驗氧化劑的選擇原則是:1)能存在于酸性介質中完全氧化次亞磷酸;2)不影響后續測定。
首先,針對第一條,即氧化劑必須在本實驗條件下具有氧化能力,開展篩選實驗。結果發現,當分別加入雙氧水、高錳酸鉀、硝酸鈰銨3種試劑與次亞磷酸配制的待測液氧化后再加入沉淀劑喹鉬檸酮,由不能沉淀變為均有明顯沉淀。上述試驗初步說明3種氧化劑均可將次亞磷酸氧化為正磷酸,具有本實驗條件下的氧化能力。但是進一步的實驗發現,過量的H2O2又會與沉淀劑發生反應,導致結果偏低或者浪費沉淀劑[9]。因此雙氧水并非理想的次亞磷酸氧化劑。高錳酸鉀能夠有效氧化次亞磷酸,將其全部氧化為正磷酸根,且過量的高錳酸鉀對于喹鉬檸酮沒有影響;但高錳酸鉀作為本實驗的氧化劑的用量難以掌握,增加操作難度,如果控制不好會產生較大誤差。
相比高錳酸鉀和雙氧水,硝酸鈰銨用于氧化次磷酸、亞磷酸氧化效果理想、操作更為簡便易行,故對于硝酸鈰銨筆者做了更深入的探究。
2.3 氧化條件探究
為了掌握硝酸鈰銨氧化次亞磷酸并與喹鉬檸酮沉淀測出磷含量的最適宜實驗條件,需要研究反應條件、氧化劑用量等對測定結果的影響。由于喹鉬檸酮重量法在沉淀過程中本身需加熱處理,而硝酸鈰銨亦需在加熱條件下體現其強氧化性,由此確定本實驗條件需加熱,故對此不再深入研究。因此本實驗重點研究硝酸鈰銨的用量的影響。
2.3.1 硝酸鈰銨的用量探究
硝酸鈰銨是一種氧化劑,在本實驗條件下可以氧化次亞磷酸根,形成五價磷化合物,反應化學方程式分別為:
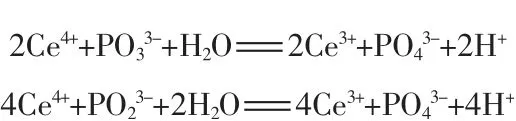
根據計算,使用本方法的硝酸鈰銨的理論用量:氧化亞磷酸需要0.1 mol/L的硝酸鈰銨7.3 mL,氧化次磷酸需要0.1 mol/L的硝酸鈰銨12.12 mL。
稱取1.000 0 g亞磷酸樣品,加入不同量、濃度為0.1 mol/L的硝酸鈰銨,對硝酸鈰銨的用量進行探究,結果見圖1。
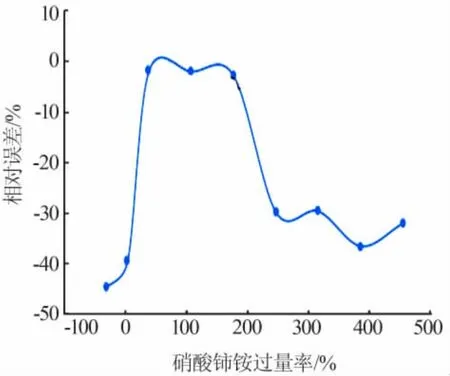
圖1 加入不同量硝酸鈰銨氧化亞磷酸Fig.1 Adding different amounts of cerium ammonium nitrate to oxidize phosphorous acid
從圖1可知,硝酸鈰銨過量率為36.99%~173.97%時相對誤差很小,超出此范圍都會導致較大的測定誤差,導致測量結果偏小。氧化劑用量不足會使次亞磷酸的氧化反應不完全而導致測量結果偏小。但氧化劑過量也會導致測得值變小并且過量越多偏離越大。對此,采用正磷酸為對象,探究加入大量的硝酸鈰銨對正磷酸的測定是否會造成影響。結果發現,在含大量正磷酸的待測液中加入大量硝酸鈰銨時會有溶液略微渾濁的現象。但同時也發現如果加入硝酸鈰銨的量不大,對正磷酸測定的影響也在合理范圍內,其結果見表2。
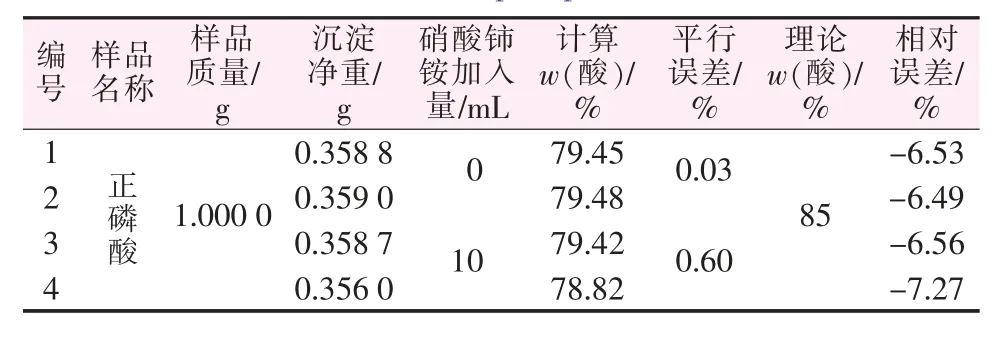
表2 硝酸鈰銨對正磷酸影響的探究Table 2 Exploration of the effect of cerium ammonium nitrate on orthophosphoric acid
綜上可知:對于未知其組成的樣品測定,硝酸鈰銨(0.1 mol/L)的實際用量應高于所計算的理論用量12.12 mL,加之硝酸鈰銨過多會對正磷酸有影響,過量的硝酸鈰銨會導致測定結果偏低,因此硝酸鈰銨也不宜過多。筆者認為硝酸鈰銨的用量在15~20 mL較合適。但實際測定情況往往不知道樣品的磷含量是多少,所以無法確定過量的合適范圍,對此還需要有進一步的解決方案。
2.3.2 異常情況及消除
由2.3.1節可知加入過量的硝酸鈰銨會導致測定結果偏小,對此筆者經實驗發現過量的硝酸鈰銨與沉淀劑喹鉬檸酮混合后會出現有少量沉淀的現象:不加待測液,于250 mL燒杯中加入100 mL去離子水,加入100 mL硝酸鈰銨,加熱至沸,再加入50 mL沉淀劑。靜置后燒杯底部有少量黃色沉淀。
理論上過量的硝酸鈰銨可能會因為產生其他沉淀而導致結果偏大,也可能會因為消耗沉淀劑而使結果偏小。結合實驗數據,總體而言其結果是沉淀劑被消耗而導致結果偏小。必須在加入喹鉬檸酮之前將過量的硝酸鈰銨除去。
硝酸鈰銨的氧化作用需要加熱,實驗發現如果在電熱板上沸騰后再加熱,過量的硝酸鈰銨會分解,故擬以加熱分解的方式除過量的硝酸鈰銨。
若在過量的硝酸鈰銨分解完全時加入少量硝酸、50 mL喹鉬檸酮,則不會再有沉淀產生干擾實驗。若加熱時間不夠長,過量的硝酸鈰銨未分解完全,則會因為與喹鉬檸酮反應而使結果極其不穩定,影響其測定。實驗結果見表3、表4。

表3 亞磷酸與硝酸鈰銨混合液加熱后測定Table 3 Determination of the mixture of phosphorous acid and cerium ammonium nitrate after heating

表4 次磷酸與硝酸鈰銨混合液加熱后測定Table 4 Determination of the mixture of hypophosphorous acid and cerium ammonium nitrate after heating
如表3、表4所示,若僅僅將待測液與硝酸鈰銨的混合溶液加熱至剛好沸騰,如表3編號1~4,測定值極其不穩定,誤差很大,但沸騰20 min可將過量的硝酸鈰銨除盡,再加入硝酸與沉淀劑,則測定值偏差更小。由此可知,溶液中過量的硝酸鈰銨必須除盡,且方法為加熱沸騰20 min以上使其分解。
3 最適宜實驗條件下對實際樣品的測定
為驗證本實驗方法的可靠性,選用不同組成的混合磷樣品、組成未知的實際磷樣品以及標準物按照本實驗方法檢驗。
3.1 混合磷樣的測定
取正磷酸、亞磷酸、次磷酸各1.000 1 g分別制成500 mL水溶液,再根據正磷酸、亞磷酸、次磷酸不同的配比配制樣品,加入0.1 mol/L的硝酸鈰銨氧化后再測定,其結果見表5~7。由表5~7可以看出,不加入氧化劑硝酸鈰銨時,因為此時僅能測出混合磷樣中正磷酸形態的磷,次磷酸、亞磷酸形態的磷沒有響應,不會形成沉淀,故混合磷樣的測定結果比真實值偏小很多。而當加入氧化劑后,沉淀包含了次亞磷酸形態的磷,才能準確反映出全部磷化合物的含量,并由此可得出次亞磷酸含量。為了進一步驗證本法,再取兩種未知組成的含磷樣品配制待測液,用本實驗方法測其磷含量,結果見表8。
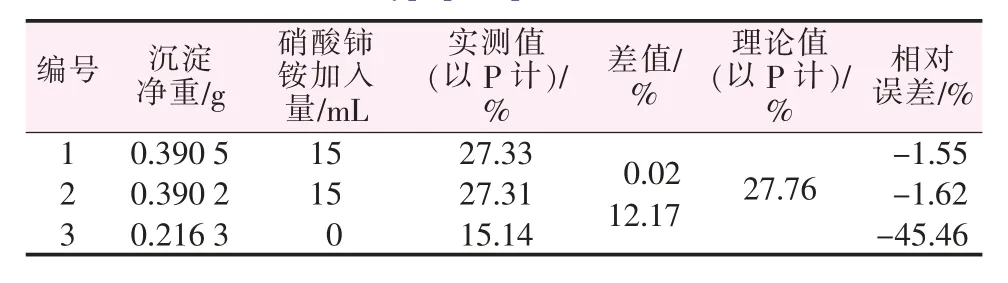
表5 含正磷酸60%、亞磷酸20%、次磷酸20%的混合樣測定結果Table 5 Determination results of mixed sample of orthophosphoric acid 60%,phosphorous acid 20%and hypophosphorous acid 20%
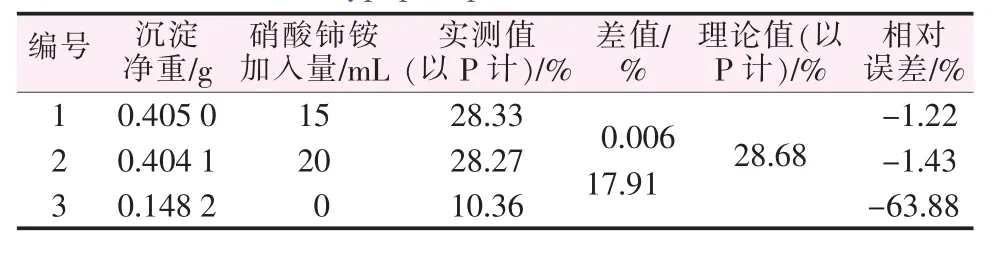
表7 含正磷酸40%、亞磷酸30%、次磷酸30%的混合樣測定結果Table 7 Determination results of mixed sample of orthophosphoric acid 40%,phosphorous acid 30%,and hypophosphorous acid 30%

表8 亞磷酸亞鐵等未知樣品的測定Table 8 Determination of unknown samples such as ferrous phosphite
硝酸鈰銨對正磷酸、亞磷酸、次磷酸含量不同試樣的氧化效果都很好且結果穩定,且由表8可知對于亞磷酸亞鐵等次亞磷酸鹽的樣品同樣適用,有實際應用的意義。
3.2 準確度試驗
選取次磷酸鈣為標準物,取1.000 1 g用本法測定,其結果見表9。由表9可知,本法相對偏差、相對誤差均<1%。
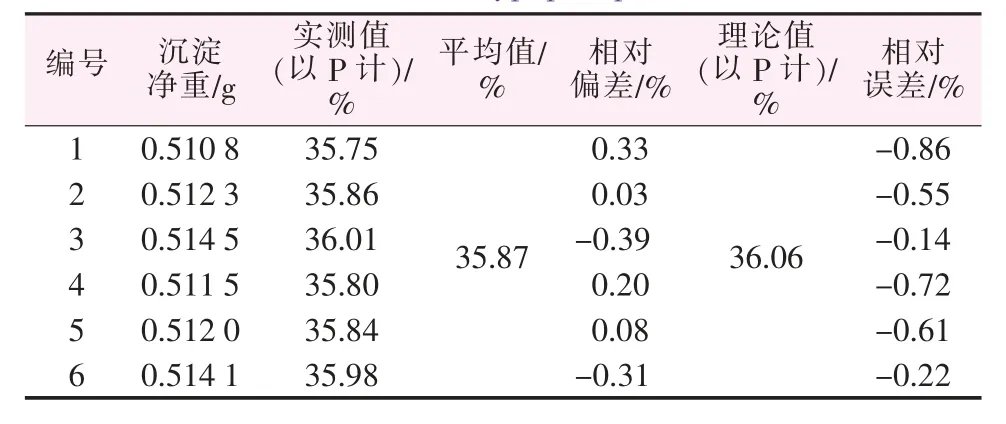
表9 對次磷酸鈣中磷含量的測定Table 9 Determination of phosphorus content in calcium hypophosphite
4 結論
1)使用傳統的喹鉬檸酮重量法無法測定低價磷組分的含量,但采用合適的氧化劑先氧化低價磷轉變為五價磷,則能夠繼續運用喹鉬檸酮重量法測定,從而解決傳統重量法無法準確測量含亞磷酸、次磷酸及其鹽等樣品的問題。
2)初步篩選出的氧化劑的選擇:高錳酸鉀、雙氧水、硝酸鈰銨3種氧化劑都能完全氧化次亞磷酸,但高錳酸鉀和雙氧水都有氧化終點無法確定的問題,且雙氧水會影響喹鉬檸酮沉淀劑,僅有硝酸鈰銨氧化劑可適用于本法。
3)實驗結果表明,使用0.1 mol/L硝酸鈰銨氧化某一待測溶液中的次磷酸和亞磷酸用量為15~20 mL為宜。無法判斷過量程度的情況下,可以加入較多的硝酸鈰銨,多余的硝酸鈰銨采用加熱至沸騰20 min以上的辦法可將過量的硝酸鈰銨除盡。
4)此法對于同時含有次亞磷酸、正磷酸的樣品,可以分別測出樣品中含有的正磷酸含量、總磷含量、次亞磷酸含量,有助于更好了解具有多形態、多組成的含磷物質的性質,更有實際應用的價值。
