大葉落地生根酵母cDNA文庫的構建及KdSAHH基因自激活檢測
2021-11-09 00:46:22王夢迪胡乃文晁躍輝曾會明
草地學報 2021年10期
關鍵詞:檢測
張 珂, 王夢迪, 董 笛, 胡乃文, 晁躍輝, 曾會明
(北京林業大學草業與草原學院,北京 100083)
大葉落地生根是景天科伽藍菜屬多年生肉質草本植物,不定芽入土即可形成新的植株,故名“落地生根”,原產于非洲馬達加斯加,廣泛分布于熱帶和亞熱帶地區[1]。大葉落地生根具有較高的研究價值,它不僅是研究景天酸代謝(Crassulacean acid metabolism,CAM)途徑的模式植物[2],也是研究高等植物無性繁殖的模式植物[3]。大葉落地生根葉緣凹陷處會形成可進行無性繁殖的不定芽,這一特質可用來研究植物體細胞全能性,為植物組織培養等相關應用的發展提供理論基礎[4]。
隨著功能基因組學研究的逐步深入,蛋白互作成為分子生物學領域的研究熱點,研究蛋白質之間的相互作用是研究基因功能的重要方法[5]。酵母雜交技術是高靈敏度的分子生物學技術,可以在酵母體內分析蛋白質互作或蛋白質與核酸互作[6]。同時,該技術既能免去純化蛋白質復雜操作且在一定程度上還能反應細胞內的真實情況[7]。酵母雜交技術已廣泛應用于蛋白質互作方面的研究,用以揭示已知蛋白或基因參與調控動植物特定代謝過程的分子機制。趙倩倩等[8]構建了玉米不同時期籽粒cDNA文庫,并從中初步篩選到了玉米籽粒灌漿期重要調控蛋白ZmSCL1的互作因子。姜紅巖等[9]探究ZjNAC2在脅迫響應中的作用機制時,成功從日本結縷草酵母雜交文庫中篩選到4個與抗病過程相關的互作蛋白。酵母雜交技術的關鍵在于構建高質量的基因文庫。因此,構建高容量的cDNA酵母文庫對酵母雜交的篩選具有重要意義。目前,對于大葉落地生根不定芽的研究集中在功能基因的克隆及表達分析方面[10-11],尚未見利用酵母雜交技術探究其分子調控機理的研究。
S-腺苷高半胱氨酸水解酶(S-adenosyl-homocysteine hydrolase,SAHH)是調節細胞內甲基化反應的關鍵酶。SAHH基因影響著植物的生長發育,抑制該基因的表達會導致植物器官的發育缺陷,如煙草花和葉的畸變[12]。本課題組[13]前期在干旱誘導的大葉落地生根不定芽差減cDNA文庫中篩選到KdSAHH基因片段,并利用熒光定量技術發現KdSAHH基因在不定芽發育過程中上調表達明顯。為探究KdSAHH基因在大葉落地生根不定芽發育過程中分子調控機制,本研究利用SMART技術構建了高容量大葉落地生根酵母雜交cDNA文庫和pGBKT7-KdSAHH誘餌載體,以期為利用酵母雜交技術從大葉落地生根cDNA文庫中篩選出KdSAHH的互作因子,揭示KdSAHH基因調控下游響應基因從而影響不定芽發生的分子機制提供研究基礎。
1 材料與方法
1.1 試驗材料
大葉落地生根培養于北京林業大學草坪研究所光照培養箱,選擇株齡為1年且不定芽生長穩定的大葉落地生根為試驗材料。RNA提取試劑盒、PCR純化試劑盒、質粒提取試劑盒均購自OMEGA公司。反轉錄試劑盒、SmaⅠ限制性內切酶、無縫連接酶均購自TaKaRa公司(日本)。文庫構建試劑盒、酵母轉化試劑盒、Y187菌株、酵母表達載體pGADT7,Y2HGold酵母菌株、酵母誘餌載體pGBKT7,X-α-Gal,AbA,Matchmaker Insert Check PCR Mix 1均購自Clontech公司。大腸桿菌感受態E.coli DH5α購自Transgen公司。
1.2 大葉落地生根總RNA的提取
選取大葉落地生根長勢良好且均一的根、莖、葉、花以及不定芽組織混勻,稱取0.1 g于液氮中研磨至粉末狀,采用試劑盒提取總RNA。用1%瓊脂糖凝膠電泳檢測總RNA的完整性,并用紫外分光光度計NanoDorp 2000測量其質量與濃度。
1.3 cDNA的合成
1.3.1cDNA第一鏈合成 取1 μg RNA為模板,與1 μL CDS Ⅲ Primer Mix混勻并加水至4 μL體系中72℃溫浴2 min,冰浴2 min后加入2 μL 5×第一鏈緩沖液,1 μL DTT,1 μL dNTP Mix,1 μL SmartscribeTM反轉錄酶,然后置于42℃保溫10 min,加入1 μL SMART III寡核苷酸,42℃保溫1 h,然后加入1 μL RNase H,37℃保溫20 min。將一鏈產物-20℃保存。
1.3.2LD PCR 取2 μL cDNA第一鏈產物和10 μL 10×Advantage 2 PCR緩沖液,2 μL 50×dNTP Mix,2 μL 5′ PCR引物,2 μL 3′ PCR引物,2 μL 50×Advantage 2 Polymerase Mix加水至100 μL體系中混勻。5′PCR引物:5′-TTCCACCCAAGCAGTGGTATCAACGCAGAGTGG-3′,3′ PCR引物:5′-GTA-TCGATGCCCACCCTCTAGAGGCCGAGGCGGCCGACA-3′。PCR反應程序為:95℃ 30 s;95℃ 15 s,68℃ 6 min,18個循環;68℃ 5 min。然后利用PCR純化試劑盒純化其產物。
1.4 cDNA文庫的均一化
1.4.1雜交 取1 μg ds cDNA,8 μL 2×雜交反應緩沖液加水至16 μL體系的PCR小管中混勻后均分4份,置于PCR儀中98℃保溫2 min后,迅速轉移到68℃水浴鍋中5 h。
1.4.2DSN消化 68℃預熱2×DSN主緩沖液。取4 μL雜交后的cDNA,5 μL預熱2×DSN主緩沖液和1 μL DSN溶液(分別為DSN,1/2 DSN,1/4 DSN,DSN儲存緩沖液)于4個PCR小管混勻,置于PCR儀中68℃保溫25 min,每管加入10 μL 2×DSN終止緩沖液后室溫放置5 min,將產物-20℃保存。
1.5 DSN消化后兩次PCR放大
1.5.1第一次放大 取5 μL 10×Advantage 2 PCR緩沖液,1 μL 50×dNTP Mix,5 μL 10×GC溶解溶液,1 μL 5′ PCR引物,1 μL 3′ PCR引物,1 μL 50×Advantage 2 Polymerase Mix和1 μL消化后產物加水至50 μL體系的PCR小管中混勻,并分別標記為DSNP1,1/2 DSNP1,1/4 DSNP1,DP1。PCR反應程序為:95℃ 30 s;95℃ 15 s,68℃ 6 min,17個循環;68℃ 5 min。用1%瓊脂糖凝膠電泳檢測后選擇模板進行第二次放大。
1.5.2第二次放大 取10 μL 10×Advantage 2 PCR緩沖液,2 μL 50×dNTP Mix,10 μL 10×GC溶解溶液,2 μL 5′ PCR引物,2 μL 3′ PCR引物,2 μL 50×Advantage 2 Polymerase Mix和2 μL稀釋十倍后一次放大產物加水至100 μL體系的PCR小管中混勻。PCR反應程序為:95℃ 30 s;95℃ 15 s,68℃ 6 min,24個循環;68℃ 5 min。用1%瓊脂糖凝膠電泳檢測,然后利用純化試劑盒純化PCR產物。
1.6 大葉落地生根cDNA酵母文庫構建與克隆鑒定
按照酵母轉化試劑盒說明書制備Y187酵母感受態,并參照趙倩倩等[8]酵母共轉化的方法轉化Y187酵母感受態細胞。吸取200 μL轉化后的酵母細胞液鋪展到規格為直徑150 mm的平板上,共計100塊平板。30℃倒置溫育平板直到克隆出現。4℃冷卻平板3~4 h,每個平板加入5 mL冷凍培養基,使用無菌的玻璃棒并輕輕的攪動,收集液體。吸取100 μL 10倍、100倍和1 000倍的酵母稀釋液鋪展到在規格為直徑100 mm SD/-Leu的平板上檢測轉化效率。文庫滴度(CFU·mL-1)=平板上的單克隆個數/平板上涂布菌液的體積(μL)×1×103(μL),文庫總容量=文庫滴度(CFU·mL-1)×文庫菌液總體積(mL)。隨機挑取24個單克隆菌落,使用Matchmaker Insert Check PCR Mix 1進行PCR檢測。上游引物序列T7:5′-TAATACGACTCACTATAGGG-3′,下游引物序列3′AD:5′-AGATGGTGCACGATGCACAG-3′。PCR反應程序為:95℃ 1 min;98℃ 10 s,55℃ 30 s,68℃ 2 min,30個循環。用1%瓊脂糖凝膠電泳檢測插入片段大小。
1.7 pGBKT7- KdSAHH載體的構建及Y2H Gold酵母轉化
根據KdSAHH基因序列(GenBank登錄號:KF953475)設計引物。上游引物序列pGBKT7-KdSAHH-F:5′-ATGGCCATGGAGGCCGAATTCG-CGCTTATCGTCGAGAAAAC-3′,下游引物序列pGBKT7-KdSAHH-R:5′-CTGCAGGTCGACGG-ATCCCCTCAGTACCTGTAGGCAGCAG-3′。提取實驗室保存測序結果正確的大腸桿菌菌液的質粒進行PCR擴增。PCR反應程序為:98℃ 10 s;98℃ 10 s,68℃ 70 s,30個循環;72℃ 5 min。pGBKT7載體用SmaⅠ單酶切后與PCR產物一同純化回收并用無縫連接酶進行載體與PCR產物的連接。將連接產物轉化至DH5α大腸桿菌感受態中,37℃倒置培養。挑取單菌落進行PCR及瓊脂糖凝膠電泳檢測,將檢測正確的菌液送至生物公司測序。提取測序正確的pGBKT7-KdSAHH重組質粒,并將其轉化至Y2HGold酵母感受態中。
1.8 誘餌蛋白自激活檢測
挑取轉化成功的酵母單菌落培養于SD/-Trp液體培養基中,30℃過夜培養,取10 μL 10倍、100倍和1 000倍酵母稀釋液滴至SD/-Trp,SD/-Trp/X-α-Gal和SD/-Trp/X-α-Gal/AbA固體培養基上,30℃倒置培養,觀察并記錄酵母生長情況和生長狀態。
2 結果與分析
2.1 大葉落地生根總RNA的提取
將提取的總RNA用1%瓊脂糖凝膠電泳檢測。結果顯示(圖1),28S和18S條帶清晰明亮,完整性好。紫外分光光度計NanoDorp 2000測量濃度為300 ng·μL-1,A260/A280值為2.11,A260/A230值為2.01,表明RNA質量好、純度高,無蛋白、酚類物質和有機溶劑等的污染,滿足建庫要求。
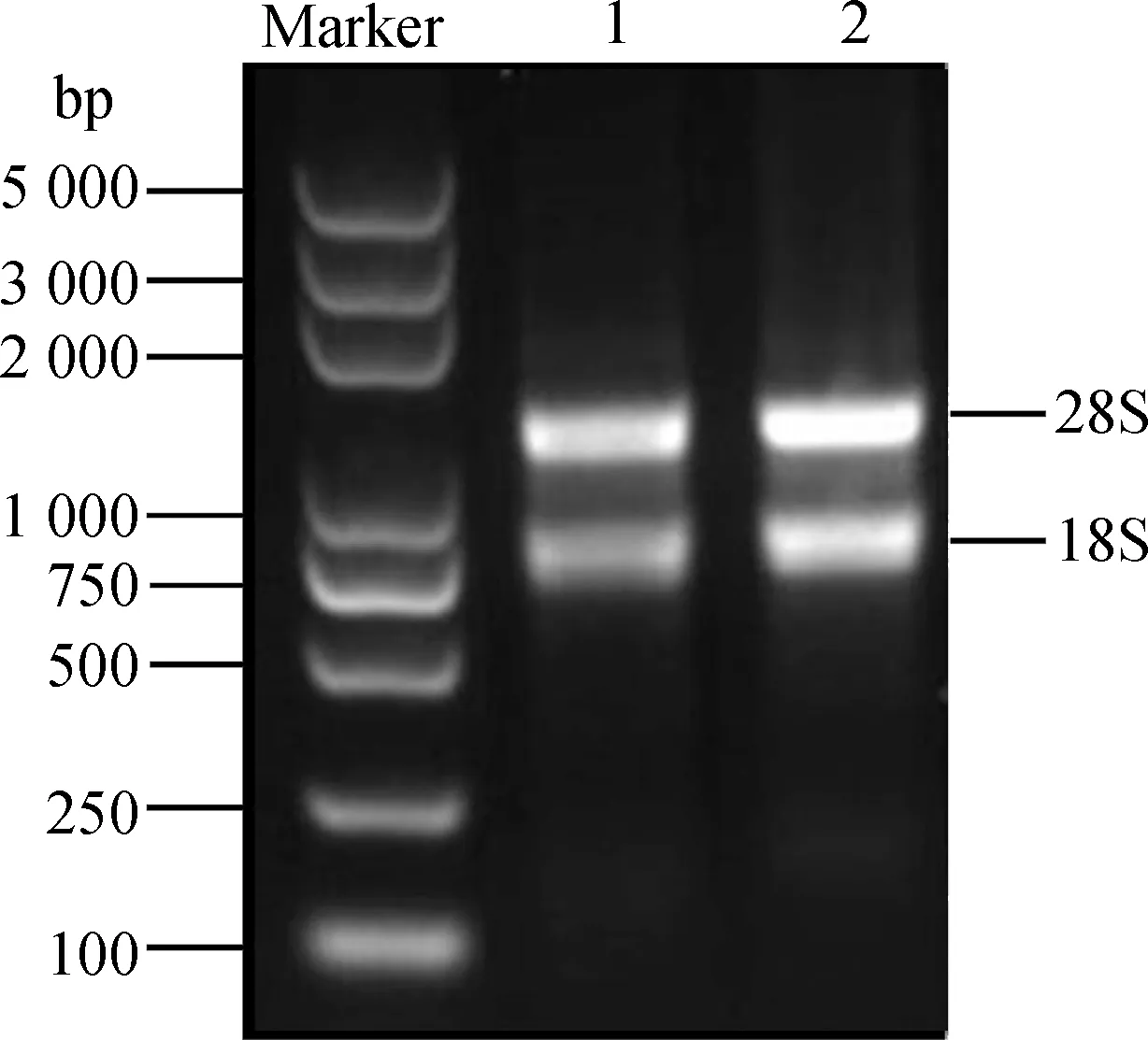
圖1 總RNA瓊脂糖凝膠電泳Fig.1 Agarose gel electrophoresis of total RNA注:Marker為Trans2K Plus DNA Marker;1和2為總RNANote:Marker indicate Trans2K Plus DNA Marker;1 and 2 indicate total RNA
2.2 雙鏈cDNA文庫的構建及均一化處理
以1 μg RNA為模板利用SMART技術反轉錄成單鏈cDNA,再以其為模板進行LD-PCR合成雙鏈cDNA。對雙鏈cDNA進行用1%瓊脂糖凝膠電泳檢測,結果顯示(圖2A),雙鏈cDNA條帶主要分布在750~3 000 bp之間,且條帶成彌散狀。將質量良好的雙鏈cDNA進行文庫均一化處理并經DSN消化后兩次PCR放大處理。結果顯示(圖2B),處理后的雙鏈cDNA條帶均一且呈彌散狀,表明均一化效果好,可用于后續試驗。
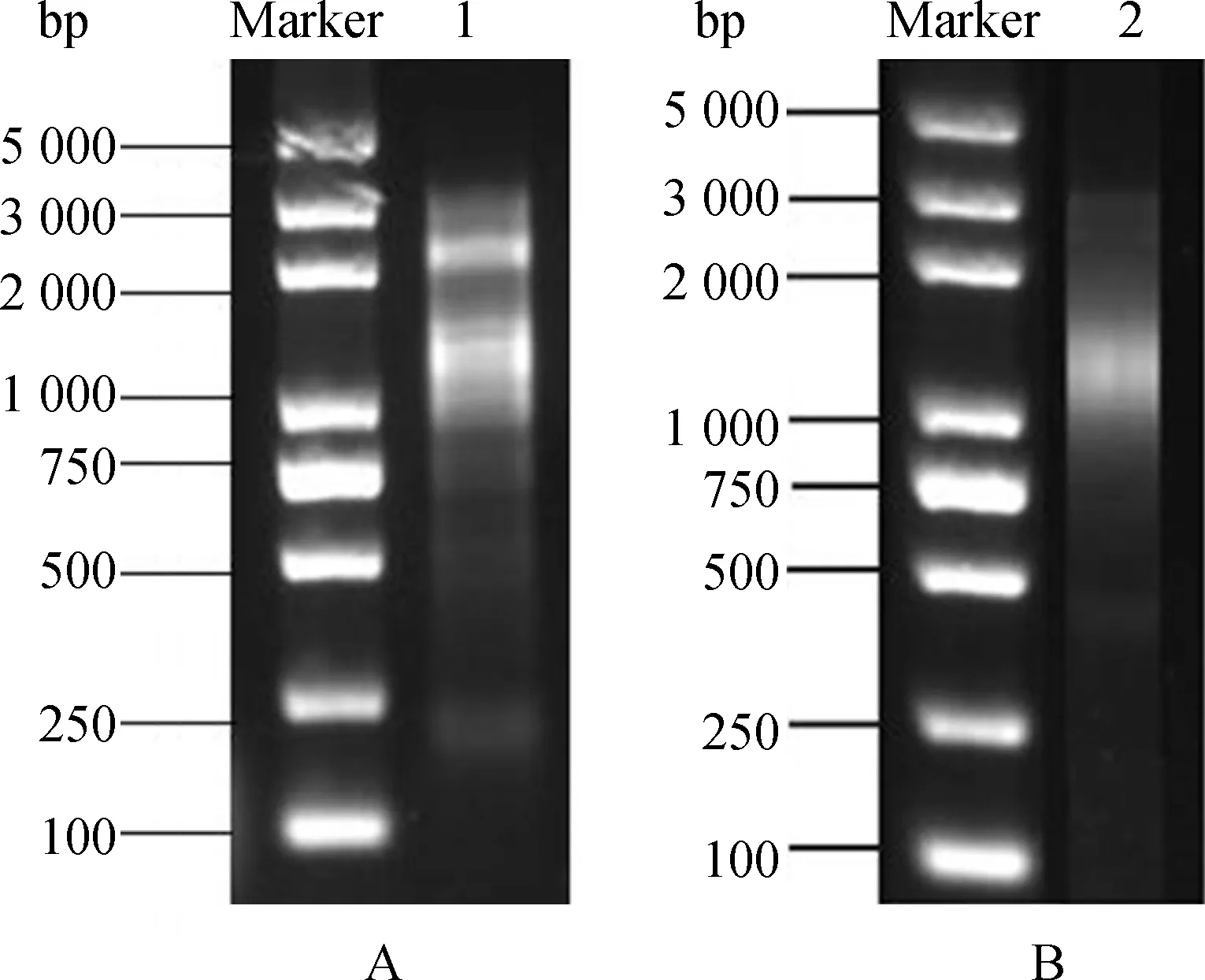
圖2 雙鏈cDNA瓊脂糖凝膠電泳(A);均一化cDNA瓊脂糖凝膠電泳(B)Fig.2 Agarose gel electrophoresis of dscDNA(A);Agarose gel electrophoresis of normalized ds cDNA(B)注:Marker為Trans2K Plus DNA Marker;1為雙鏈cDNA;2為均一化cDNANote:Marker indicate Trans2K Plus DNA Marker;1 indicate ds cDNA;2 indicate normalized ds cDNA
2.3 大葉落地生根cDNA酵母文庫構建與質量檢測
將ds cDNA轉化至提前制備好的Y187酵母感受態細胞中。菌液稀釋液鋪展在100 mm SD/-Leu平板上倒置培養3~4 d后,在1 000倍稀釋液的平板上大約有酵母單菌落320個(圖3A)。根據文庫滴度和文庫總容量公式可以得出所構建的文庫滴度為3.2×106CFU·mL-1,文庫總容量為4.8×107CFU。
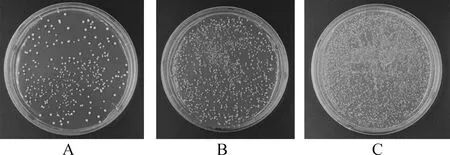
圖3 不同濃度文庫菌液的單菌落Fig.3 The clone of different concentrations library suspension注:A為稀釋1 000倍的菌液;B為稀釋100倍的菌液;C為稀釋10倍的菌液Note:A indicate 10-3 library dilution;B indicate 10-2 library dilution;C indicate 10-1 library dilution
從100 mm SD/-Leu平板上隨機挑取24個酵母單菌落進行PCR擴增檢測隨機插入片段長度。電泳結果顯示(圖4),重組率為91.7%,24個單克隆中除第10和23無條帶外其余均有清晰的單一條帶,且擴增條帶大小不一,插入片段長度在250~2 000 bp范圍內。
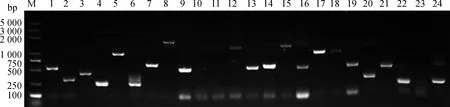
圖4 大葉落地生根cDNA酵母文庫插入片段大小檢測Fig.4 The insertion size range of yeast cDNA library of Kalanchoe daigremontiana注:Marker為Trans2K Plus DNA Marker;1~24為擴增產物Note:M indicate Trans2K Plus DNA Marker;1~24 indicate amplifications products
2.4 誘餌載體pGBKT7-KdSAHH構建及酵母轉化
將已完成PCR擴增的KdSAHH片段與酶切后的pGBKT7誘餌載體純化連接,然后將連接產物轉化至DH5α大腸桿菌中。挑取單菌落搖菌,然后進行菌液PCR鑒定及瓊脂糖凝膠電泳檢測(圖5),1 800 bp附近出現清晰條帶,將該菌液送至生物公司測序。返還測序結果正確,誘餌載體pGBKT7-KdSAHH構建成功。提取測序正確的pGBKT7-KdSAHH重組質粒,并將其轉化至Y2HGold酵母感受態。取100 μL菌液均勻鋪展到SD/-Trp培養基上,30℃培養2~3 d。菌落能正常在營養缺陷培養基上生長,說明表達蛋白對酵母無毒害作用。
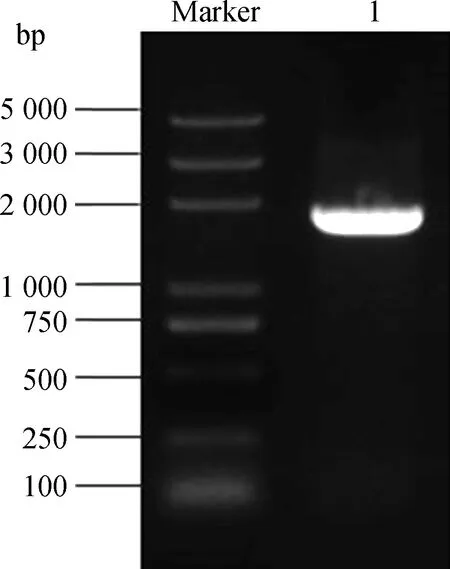
圖5 誘餌載體瓊脂糖凝膠電泳Fig.5 Agarose gel electrophoresis of bait vector注:Marker為Trans2K Plus DNA Marker;1為誘餌載體Note:Marker indicate Trans2K Plus DNA Marker;1 indicate bait vector
2.5 載體轉錄自激活檢測
通過觀察酵母單菌落在營養缺陷型培養基上的生長情況和生長狀態進行轉錄自激活檢測。酵母菌在SD/-Trp固體培養基上正常生長,在SD/-Trp/X-α-Gal固體培養基上正常生長且不顯藍色,在SD/-Trp/X-α-Gal/AbA固體培養基上不能正常生長且不顯藍色(圖6)。載體轉錄自激活檢測表明,KdSAHH不具備轉錄自激活活性,可以進行下一步酵母雜交試驗篩選互作蛋白。
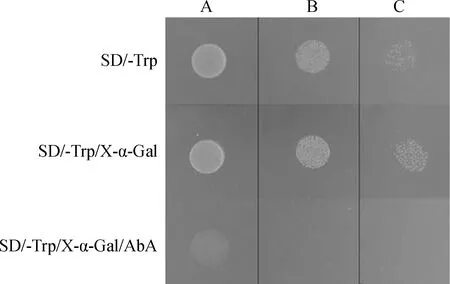
圖6 pGBKT7-KdSAHH載體轉錄自激活檢測Fig.6 Self-activation detection of pGBKT7-KdSAHH注:A為稀釋1 000倍的菌液;B為稀釋100倍的菌液;C為稀釋10倍的菌液Note:A indicate 10-3 library dilution;B indicate 10-2 library dilution;C indicate 10-1 library dilution
3 討論與結論
cDNA文庫已廣泛應用于生物技術領域的各項研究,因此構建高容量的cDNA文庫是進行有效研究的重要基礎[14]。文庫質量主要從文庫代表性及重組序列的完整性這兩個指標進行評價[15-16]。本研究建庫材料來源于大葉落地生根各個植物組織,為cDNA種類的完整性提供保障。經過文庫菌液稀釋涂板檢測(圖3A),文庫滴度為3.2×106CFU·mL-1,達到低豐度mRNA篩選要求(>1×106CFU·mL-1),文庫總容量為4.8×107CFU,由此可見,所建文庫含有的遺傳信息較豐富,文庫代表性好。隨機挑取24個酵母單菌落進行PCR擴增(圖4),檢測隨機插入片段長度在250~2 000 bp范圍內,文庫多態性好,重組率為91.7%。同時,通過觀察含有pGBKT7-KdSAHH載體的酵母菌在營養缺陷型培養基上的生長情況和生長狀態,證明KdSAHH不具備轉錄自激活活性,可用于后續酵母雙雜交試驗。
本研究采用由美國Clontech公司推出的SMART技術[17]和DSN技術[18]構建大葉落地生根全長cDNA文庫。將SMART技術所需RNA少、操作步驟少、用時較短且獲得的文庫全長比例高等優點與DSN酶的選擇性降解雙鏈核酸分子中的DNA這一特點結合起來,解決了建庫過程中mRNA純化效果不好且低豐度表達的基因易丟失的問題[19],同時還增加篩到稀有基因的可能性。但用上述方法構建的文庫中有個別克隆片段偏小,插入片段的平均長度略小于1 000 bp。韋春艷等[20]在構建陸地棉酵母雙雜交cDNA文庫時,文庫插入片段同樣偏小,但并未影響后續試驗的開展。總體來說,本試驗構建的大葉落地生根cDNA酵母文庫質量較高,滿足后續酵母雜交試驗的要求。
目前,在大葉落地生根不定芽發生過程中已經鑒定出一些重要調控蛋白[21-22],但具體的信號轉導途徑與分子調控網絡尚不清楚。以已知調控蛋白為誘餌,構建誘餌表達載體,通過酵母雜交技術在大葉落地生根cDNA酵母文庫篩選互作蛋白,該研究思路為探究大葉落地生根基因功能及功能基因的調控網絡提供幫助。本研究成功構建了高容量的大葉落地生根cDNA酵母文庫和pGBKT7-KdSAHH誘餌載體,轉錄自激活檢測表明KdSAHH無轉錄自激活活性。接下來利用酵母雜交技術進行KdSAHH互作驗證等試驗的開展將為揭示KdSAHH基因調控下游響應基因從而影響不定芽發生的分子機制奠定研究基礎。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
