
尼曼-匹克病C2型一例報道并文獻復習
2021-12-07 10:36:44毛春婷楊軍時珺繆紅軍李軍
中國全科醫學 2021年3期
毛春婷,楊軍,時珺,繆紅軍,李軍
尼曼-匹克病(NP)是一組常染色體隱性遺傳性溶酶體內脂質貯積病,主要分為A、B、C型,目前研究最多的是NP C 型。NP C 型發病率為 1:100 000~1:150 000[1],其中僅5%為NP C2型,因此NP C2型非常罕見[2]。現報道南京醫科大學附屬兒童醫院急診科最近收治1例2歲8個月NP C2型女性患兒,并進行相關文獻復習,以提高兒科醫生對該疾病的診治水平,減少漏診、誤診。
本文價值:
小兒最常見呼吸系統和消化系統疾病,且臨床對于以雙肺彌散性病變為突出表現,合并肝脾腫大、精神運動發育落后的尼曼-匹克病C2型(NP C2)極易誤診、漏診。本例患兒出現了遷延不愈的肺部病變、慢性肝脾腫大和語言、認知及運動發育落后等表現,作者本著盡量用一個疾病解釋所有癥狀的原則,總結臨床表現的關鍵詞,檢索文獻尋找相應的疾病,最終考慮為NP C2,并進行基因檢測使診斷明確,解釋了患兒看似不相關的復雜表現的最終病因,該診斷思路為臨床醫生提供了好的典范。另外,本例患兒的纖維支氣管鏡病理活檢結果提示“肺泡蛋白沉積癥”,但作者并未停留于此診斷,而是追根溯源從NP C2的基因變異和蛋白質翻譯異常的角度解釋了這一現象,這種“知其然,知其所以然”的科研精神也值得提倡。
1 病例簡介
患兒,女,2歲8個月,因“反復咳嗽2年余,加重伴發熱、氣促半天”入住南京市兒童醫院急診觀察室。入院查體:體溫38.2 ℃,心率153次/min,呼吸52次/min,體質量6.8 kg。鼻導管吸氧,意識清、精神萎,反應遲鈍,不會說話,皮下脂肪菲薄,未見皮疹,淺表淋巴結未及腫大。呼吸急促,有胸骨上窩吸氣性凹陷,面色蒼白,口唇微發紺,咽部稍充血,頸軟,雙肺呼吸音粗,可聞及大量中細濕啰音和喘鳴音,心音稍鈍,心律齊,未聞及病理性雜音,上腹部及左側胸壁可見兩處陳舊性手術瘢痕,腹軟,肝肋下約20 mm可及,質中。脾肋下約35 mm可及,質中。全腹未觸及包塊。四肢肌張力低下,四肢肌力2~3級,共濟失調,不能翻身、獨坐。雙側巴氏征陰性。
患兒系第一胎第一產(G1P1),足月剖宮產,出生體質量3.0 kg。患兒生后即發現皮膚黃疸,且大便呈淡黃色,出生后第2個月在上海復旦大學附屬兒科醫院診斷為膽汁淤積,并行手術治療。
患兒生后不足1個月開始出現反復咳嗽,胸部CT提示雙肺彌漫性病變,多次呼吸道感染后出現咳嗽加重,有明顯氣促、口唇發紺,抗感染、平喘、吸痰等治療后咳嗽有緩解,曾在南京市兒童醫院多次住院治療,有呼吸窘迫入住兒童重癥病房監護室(PICU)史。出院時仍病因不明確,隨訪2年胸部CT仍提示雙肺彌漫性病變。2018年3月于南京市兒童醫院胸外科行全麻胸腔鏡下左下肺葉楔形切除術,術中探查見“左側肺葉,色蒼白,彈性差,肺表面呈顆粒狀”,術后病理結果提示肺泡蛋白沉積癥。2018年5月于呼吸科行支氣管鏡檢查見雙側支氣管黏膜稍充血,右上、中、下葉支氣管開口及左上、下支氣管開口見白色黏稠分泌物,予負壓吸引后通暢,各葉段支氣管開口未見異物及新生物堵塞。2018年5月出院后,居家吸氧、口服阿奇霉素維持治療。
實驗室檢查:血常規:C反應蛋白(CRP)13 mg/L,白細胞計數13.9×109/L,中性粒細胞百分數77.5%,血紅蛋白142 g/L,血小板計數234×109/L,降鈣素原2.07 μg/L(參考范圍<0.05 μg/L);生化檢查:丙氨酸氨基轉移酶8 U/L,天冬氨酸氨基轉移酶126 U/L(參考范圍0~40 U/L),乳酸脫氫酶607 U/L(參考范圍80~285 U/L),肌酸激酶同工酶22 U/L,總蛋白66.6 g/L,白蛋白45.7 g/L,總膽紅素2.84 μmol/L,尿素3.44 mmol/L,肌酐15.5 μmol/L;呼吸道九項:肺炎支原體免疫球蛋白M(IgM)(弱陽性),余陰性;淋巴細胞免疫分析:自然殺傷細胞(NK細胞)3.83%(參考范圍6%~27%),B淋巴細胞27.76%;體液免疫5項、甲狀腺功能7項、自身抗體全套、血液串聯質譜、尿氣相質譜均未見異常。心電圖檢查示竇性心動過速。腹部B超示,肝:右葉斜徑63 mm,肋下20 mm,形態規則,光點均勻;脾臟:肋間厚24 mm,肋下33 mm,形態規則,脾內光點均勻分布。顱腦1.5 T磁共振成像(MRI)平掃+彌散成像:雙側大腦半球對稱,雙側額顳頂葉腦溝增寬加深,腦回形態無異常;胼胝體變薄、信號正常;腦干、小腦形態、信號未見明顯異常;垂體信號可見腦萎縮樣改變(見圖1)。氣管及支氣管1.5 T MRI平掃+重建:胸廓欠對稱,雙肺野透亮度欠對稱,雙肺紋理增多、模糊,雙肺示大片狀磨玻璃樣影及斑片狀致密影,肺血管增粗,余肺野透亮度增高;胸腔內未見積液;縱隔內及左腋下可見片狀鈣化影;提示雙肺彌漫性病變;縱隔內鈣化影,左腋窩淋巴結鈣化(見圖2);氣管支氣管重建未見明顯異常;雙肺彌漫性病變(2018-03-20)改變不明顯。2018-08-15骨髓穿刺病理報告結果:閱片可見類似NP細胞(見圖3)。
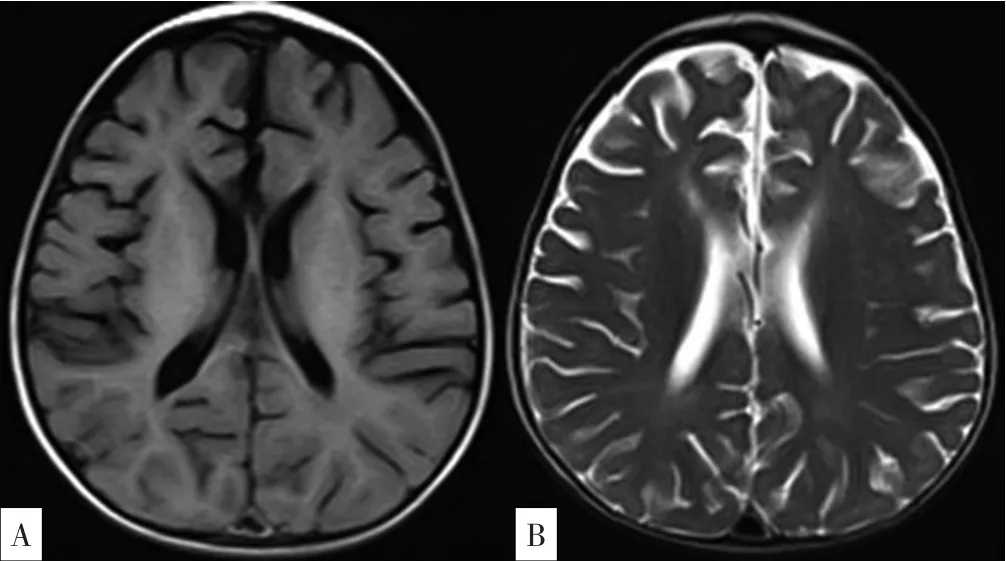
圖1 患兒顱腦MRI檢查結果Figure 1 Results of brain MRI in this case
2018年8月行基因測序發現,受檢者NPC2基因有1個純合突變:c.258delC(缺失胞嘧啶),導致氨基酸改變p.187Ffs*24(移碼突變);次突變為新發突變,在人類基因突變數據庫(HGMD)未見報道,受檢者之父、母均有chrl4-74951222-74951223位點雜合突變(見圖4),提示疾病:NP C2型。
2 文獻復習
以“Niemann-Pick disease type C2”為檢索詞,檢索起止時間為建庫至2018年9月,檢索PubMed,發現10篇NP C2病例報道[3-12],均為英文報道(有2篇文獻報道不同時期同一例患兒,1篇為產前診斷),共有8例患者,其共同的、最突出的臨床表現為呼吸窘迫(8/8),其中7例患兒在13個月前因呼吸衰竭死亡;1例患兒于16個月時接受骨髓細胞移植治療,隨訪至5歲,其言語障礙方面有改善,但大動作發育落后及肺部病變改善不明顯。有4例患兒存在肌張力低下、共濟失調、精神發育遲滯及顱腦MRI檢查異常,其余4例因死亡時間太早(<4個月),文章中未明確報道神經系統異常。
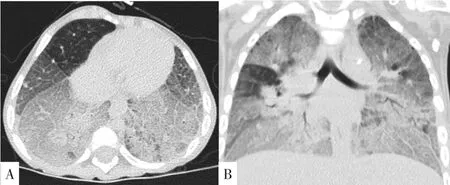
圖2 患兒氣管及支氣管MRI檢查結果Figure 2 MRI findings of trachea and bronchus in this case
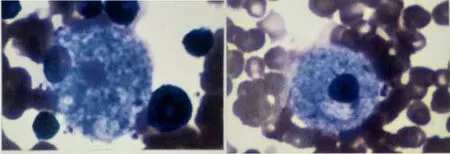
圖3 患兒骨髓穿刺病理檢查結果(瑞氏-姬姆薩染色,×1 000)Figure 3 Pathological results of bone marrow puncture in this case
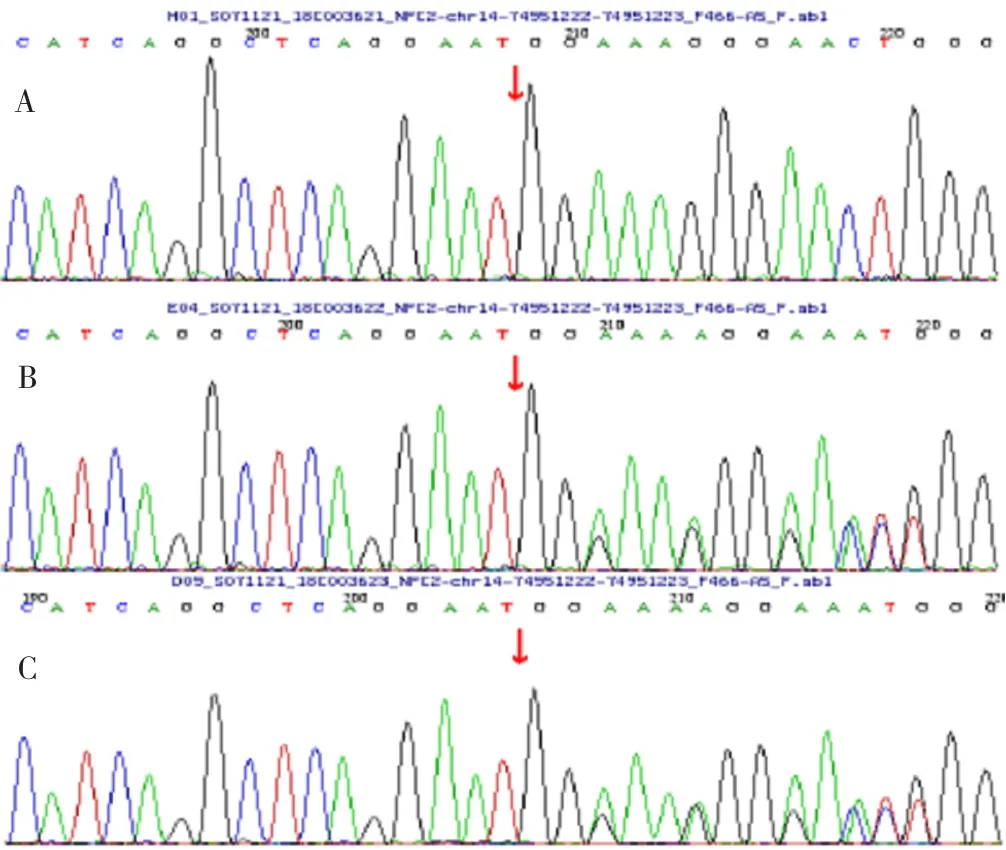
圖4 患兒及其父母NPC2基因測序結果Figure 4 Sequencing of the gene associated with NPD type C2 in this case and her parents
以“尼曼匹克病”為主題詞,再次檢索中文數據庫(中國知網、萬方數據庫),檢索起止時間為建庫至2018年9月,篩選條件為年齡<18歲,最終納入31篇病例報道,共65例患兒。患兒中男42例,女23例,男∶女為1.83∶1;發病年齡2 d~17歲。基因診斷為NP C1型28例、A型1例,臨床診斷為A型4例、B型2例、C型1例、E型1例,其余28例骨髓病理檢測到NP細胞,結合臨床表現診斷,未分型。63例(95.4%)患兒存在肝/脾腫大,52例(80.0%)存在營養不良,53例(81.5%)存在精神運動發育落后,55例(84.6%)存在肌張力低下,28例(43.1%)存在嬰兒期嚴重黃疸病史,4例(6.2%)存在癲癇、猝倒,2例(3.1%)存在眼球垂直運動障礙,2例(3.1%)存在眼底櫻桃紅斑,2例(3.1%)存在腎病表現。19例(29.2%)患兒有反復肺部感染病史,其中28例未分型患兒中有6例[13-16]以反復嚴重的肺部感染為主要臨床表現,肺部影像學檢查提示肺間質病變、雙肺彌散性病變、肺部粟粒樣病變。這6例患兒發病年齡小(2 d~3歲),其中3例分別于2 d、40 d和10個月因呼吸衰竭死亡,尸檢顯示肺、肝、腦部見NP泡沫細胞,其中10月齡患兒肺部散在0.5~1.5 cm脂質黃斑。這6例患兒的臨床表現與國外報道的NPC2型患兒非常類似,但遺憾的是這些病例未進行基因測序。
3 討論
NP是1914年首先由德國醫生Niemann報告,1922年Pick詳細描述了病理檢查所見,故而得名[1]。1961年Crocker將NP分為4型:A型為急性嬰兒型,患兒重度肝脾腫大伴神經系統異常;B型為青少年-成人型,僅內臟器官受累;C型和D型臨床特征相似,嬰兒至成年人均有發病,臨床表現多樣,包括一系列神經精神癥狀,肝、脾和肺部病變,現D型已不再是獨立的疾病。國內曾有學者提出將發病年齡晚(大多數為成年期)、無神經系統表現者歸為E型[17]。A型和B型NP的發病機制是編碼溶酶體酸性神經鞘磷脂酶基因突變,致多器官細胞溶酶體內進行性神經鞘磷脂和其他脂質貯積[17]。目前病例報道最多、研究最深入的是C型。
NP C型患兒的臨床表現多種多樣,不同發病年齡有不同臨床表現。發病年齡為0~2個月的NP C型患兒臨床表現主要以肝病為主,膽汁淤積性黃疸、肝脾輕度腫大。8%~9%的NP C型患兒有急性進行性肝衰竭,這部分患兒一般在6月齡前死亡[18]。2月齡~2歲發病的患兒有生長遲緩、精神運動發育遲滯、肌張力低下等神經系統表現,部分患兒有肺部疾病表現[18]。許多疾病如特發性肝炎、膽管閉鎖、Wolman病、Gaucher病Ⅲ型等均存在嬰兒期長時間黃疸、肝脾腫大等臨床表現,應注意與NP C型鑒別,鑒別要點在于以上疾病基本不伴隨早發的發育遲滯。2~6歲發病的NP C型患兒臨床表現主要為運動機能損傷,如笨拙、步態紊亂、精細運動障礙、垂直核上凝視麻痹(VSGP)、言語延遲等。6~15歲NP C型青少年發病的臨床表現常為認知損害、學習困難、運動協調障礙等。>15歲NP C型發病者臨床表現常為認知損害,并趨向于呈現精神疾病的神經系統表現。因此,NP C型臨床誤診率高,但是如果VSGP特征被識別,可減小誤診率[19]。
NP C型是一種獨特的、胞內、外源性膽固醇和多種脂質轉運異常性疾病,主要由NP C1或NP C2基因突變所致,NP C1或NP C2兩種蛋白質功能障礙,未酯化的膽固醇在細胞中逐漸累積,導致細胞死亡和器官損傷[18,20]。95%的NP C型病例是內涵體膜蛋白NP C1基因突變所致的NPC1型。NP C1基因位于染色體18q11-12,約47 kDa。NP C1蛋白能感知胞吞進入細胞的膽固醇,于膽固醇升高時進入或存留于次級內涵體、溶酶體膜,結合并轉運膽固醇。5%的NP C2型病例源于NP C2基因突變。NP C2基因位于染色體14q24.3,編碼151個氨基酸組成的可溶性糖蛋白,分子量小(16 kDa),主要存在于次級內涵體/溶酶體,由甘露糖-6-磷酸途徑分泌和再捕獲,在各種組織均有表達。NP C2基因編碼的NPC2蛋白具有由3個二硫鍵穩定的Ig樣折疊;疏水核的疏松填充區域,形成相鄰的小空腔,成為膽固醇的結合位點。NP C2蛋白具有高度可塑性,其表面有幾個強烈的帶正電區域,這些區域可以與帶負電的膜磷脂發生良好的相互作用[21]。因此,NP C2蛋白以1∶1的化學計量比直接結合膽固醇,通過蛋白質-膜相互作用的機制在次級內涵體/溶酶體蛋白質的多層緊密貼合的膜之間快速運輸膽固醇,將胞內膽固醇轉運至胞外,并在調節胞內膽固醇穩態中發揮重要作用。當胞內膽固醇穩態的調節機制障礙,膽固醇持續積聚,溶酶體/次級內涵體不斷增大,就形成典型的菲律賓染色陽性的泡沫狀NP細胞。
NP C1型通常以肝脾腫大和嚴重的進行性神經功能障礙為臨床特征,而累及肺部者并不多見,但NP C2型患兒通常在嬰兒期出現呼吸窘迫,合并肺泡蛋白沉積癥。RAMIREZ等[22]報道NP C1基因敲除小鼠的肺重量、膽固醇和磷脂(PL)水平以及膽固醇合成速率均升高,肺三酰甘油水平降低,肺泡中載脂巨噬細胞增加;在NP C2基因敲除小鼠的肺中,類似的代謝和組織學改變比NP C1基因敲除小鼠更加明顯。NP C2外顯子4移碼突變c.408_409delAA使血清和肺灌洗液中的NP C2蛋白水平降低,肺泡巨噬細胞的蛋白表達也顯著減少,但富含膽固醇的功能性非活性表面活性劑(約28 kDa的異常大蛋白)合成增加,臨床表現為肺泡蛋白沉積癥、嬰兒肺發育不良、呼吸窘迫,影像學結果顯示雙肺彌散性病變[23]。因此,不管是病例特點,還是動物實驗均提示早期出現呼吸窘迫和肺泡蛋白沉積癥是NP C2型突出的臨床特征。
一旦臨床懷疑NP C型,可通過生物化學檢測和分子遺傳學分析進行確診。近年來,幾種血漿代謝物膽甾醇-3β,5α,6β-三醇溶血磷脂鞘磷脂異構體和膽汁酸代謝物已成為PN C型特異性生物診斷標志物。但基因檢查仍被認為是確診NP的一線實驗室檢查結果。從本例患兒和文獻復習資料來看,骨髓穿刺發現泡沫狀尼曼匹克細胞是簡便快捷而準確的輔助診斷方法。PN C型的顱腦影像學檢查是非特異性的,但最常見的報告是小腦、海馬和皮質下灰質體積減少,白質區可見細微異常。本例患兒的顱腦MRI示腦萎縮改變。過去PN C型無有效的治療方法,但隨著米格盧斯塔特(miglustat)的研發成功,目前建議一旦確診PNC,即予米格盧斯塔特治療[24]。米格盧斯塔特是唯一的底物減少療法,也是歐洲藥品管理局批準的治療神經系統疾病的改良藥物,且已被證明在臨床可終止或減緩NP C型的病情進展[24]。另外,鞘內注射或靜脈注射2-羥丙基-β-環糊精和口服阿莫洛莫的安全性和有效性的臨床試驗還在進行中。雖然NP存在膽固醇轉運紊亂,但降低膽固醇的藥物尚未被證明在改變NP病情方面有效。
綜上所述,本例患兒系NP C2型,臨床非常罕見,目前為我國首例報道。呼吸窘迫是NP C2型最突出的臨床表現,對于不明原因的反復呼吸道感染合并精神運動發育落后、肝脾腫大者,應警惕NP的存在。
作者貢獻:毛春婷負責文章的構思與設計、病例資料收集和論文撰寫;楊軍、時珺、繆紅軍負責修改論文;李軍進行文章的質量控制及審校,并對文章負責。
本文無利益沖突。
