珠海市不同時期新型冠狀病毒基因組測序及溯源分析
2021-12-15 07:56:00龍冬玲黃輝濤魏泉德黃文燕
中國人獸共患病學報 2021年11期
龍冬玲,黃輝濤,魏泉德,黃文燕
2019年12月,新型冠狀病毒(SARS-CoV-2)在武漢被首次報道,目前該病毒在全球范圍內(nèi)已造成大流行[1]。截至北京時間2021年5月6日6時30分,全球累計確診新冠肺炎病例155 790 320例,累計死亡3 254 439例,全球單日新增確診病例857 854例[2]。新型冠狀病毒屬于β屬冠狀病毒,是迄今為止被發(fā)現(xiàn)的冠狀病毒中第7種可以使人類致病的冠狀病毒,其全基因組包含近30 000個核苷酸堿基,由14個蛋白編碼區(qū)域構成。目前研究顯示,新型冠狀病毒與蝙蝠類SARS樣冠狀病毒基因組相似性最高,與導致人類感染的其他冠狀病毒差異較大。新型冠狀病毒全基因組測序可提示病毒的變異情況和揭示傳染病的傳播途徑,對疾病防控策略的制定具有重要意義。珠海市于2020年1月19日檢出首例新型冠狀病毒肺炎病例,截止至2021年5月5日24時,全市累計確證病例113例,其中15例為境外輸入病例[3]。為了解珠海市新型冠狀病毒的變異和溯源情況,為后期及時準確追蹤溯源病毒,我們選取了4份不同時期確診為新型冠狀肺炎病例的呼吸道標本進行測序分析,現(xiàn)將結果分析如下。
1 材料與方法
1.1 樣本來源 4份上呼吸道樣本均采集于珠海市2020年1月至12月確診為SARS-CoV-2病例患者標本,其中咽拭子樣本3份,鼻咽拭子1份。采用中山達安基因股份有限公司的新型冠狀病毒2019-nCoV核酸檢測試劑盒檢測,確診病例疾病分型參照新型冠狀病毒肺炎診療方案(試行第七版)[4],具體樣本信息見表1。
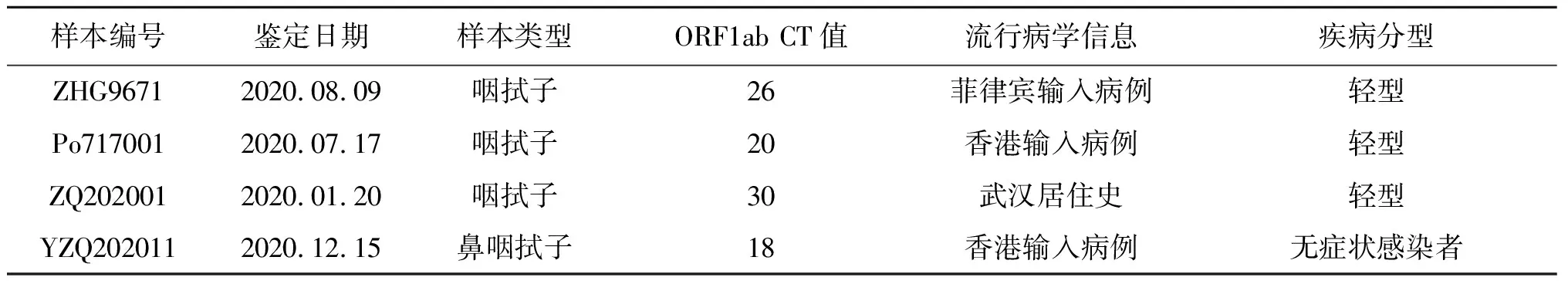
表1 標本信息Tab.1 Sample information
1.2 全基因組文庫構建和測定 采用病毒核酸提取試劑盒(Qiagen RNeasy Mini Kit,貨號:74104)提取200 μL樣本的總RNA;應用隨機引物逆轉錄方法,采用逆轉錄試劑盒(LunaScriptTMRT SuperMix Kit,貨號:E3010L)生成cDNA。根據(jù)ARTIC Network公布的實驗方案合成擴增引物(COVID-19 primers V3版)。采用多重PCR擴增試劑盒(Q5○RHot Start High-Fidelity 2X Master Mix,貨號:M0494S)獲得多重擴增產(chǎn)物。反應條件:98 ℃ 30 s(1 cycle);98 ℃ 15 s,65 ℃ 5 min(35 cycle);65 ℃ 5 min(1 cycle)。擴增產(chǎn)物回收采用核酸磁珠純化試劑盒(AMPure XP beads,貨號:A63880)等比例純化回收。使用核酸定量試劑(Qubit dsDNA HS ASSAY kit,貨號:1646715)對純化核酸進行定量。采用產(chǎn)物末端修復試劑盒(NEB Next End repair/dA-tailing Module,貨號:E7546S)對擴增產(chǎn)物末端修復,接頭連接采用試劑盒(NEB Next Quick Ligation Module,貨號E6056S;Native Barcoding Kit,EXP-NBD104/EXP-NBD114)對樣本進行無擴增條形碼標記與接頭連接。上機測序采用試劑盒(Flow Cell Primming Kit,貨號:EXP-FLP002)進行高通量測序。
1.3 數(shù)據(jù)分析處理 使用RAMPART軟件對下機數(shù)據(jù)進行序列修剪和質(zhì)量評估,使用minimap2和medaka組裝軟件生成一致性序列。參考基因組為早期公布的2019-nCoV病毒株Wuhan-Hu-1的基因組(GenBank Accession No.NC045512.2)。采用IQ-TREE軟件構建序列分子進化樹,進化樹構建所納入的參考序列以在GISAID數(shù)據(jù)庫中篩選出來與所測序列相似性最高、同時期的國際上公布的序列作為同源性分析的參考基因組,每條目標序列選取250條相似序列進行分析,相同的參考基因組只選擇一次,相似性序列查找完成后使用mega7.0軟件線下分析,最大似然法構建系統(tǒng)進化樹,步長值選擇1 000,納入線下分析的參考基因組分別為hCoV-19/Wuhan/WH01/2019、hCoV-19/Colombia/AMA-INS-VG-1193/2021、 hCoV-19/England/20134017404/2020、 hCoV-19/England/CAMC-137AA76/2021、hCoV-19/England/QEUH-C5B84E/2020、 hCoV-19/Guangdong/20SF028/2020、hCoV-19/Guangdong/20SF040/2020、hCoV-19/HongKong/HKPU-01524/2020、 hCoV-19/HongKong/HKPU-01574/2020、hCoV-19/HongKong/HKPU-02039/2020、hCoV-19/HongKong/HKPU-02290/2020、hCoV-19/HongKong/HKPU-06284/2020、hCoV-19/HongKong/HKPU-07782/2020、hCoV-19/HongKong/HKPU-08506/2020、hCoV-19/HongKong/HKPU-5161/2020、 hCoV-19/HongKong/HKPU-04778/2020、hCoV-19/HongKong/HKPU-5183/2020、hCoV-19/HongKong/HKU-201216-036/2020、hCoV-19/HongKong/HKU-201216-037/2020、hCoV-19/HongKong/HKU-201216-060/2020、hCoV-19/HongKong/HKU-201216-172/2020、hCoV-19/HongKong/HKU-201216-232/2020、hCoV-19/HongKong/HKU-201216-260/2020、hCoV-19/HongKong/HKU-201216-346/2020、hCoV-19/HongKong/HKU-201216-367/2020、hCoV-19/HongKong/HKU-201216-372/2020、hCoV-19/HongKong/VM20040795/2020、hCoV-19/Spain/NC-IBV-001418/2020、hCoV-19/Thailand/Chon-buri-SQ-NP0073/2020、hCoV-19/USA/NC-CDC-STM-000022772/2021。
2 結 果
2.1 基因組測序基本情況 4份樣本經(jīng)nanopore三代基因組測序結果見表2,基因組平均測序深度3 468×,基因組覆蓋度在94.6%~99.7%。
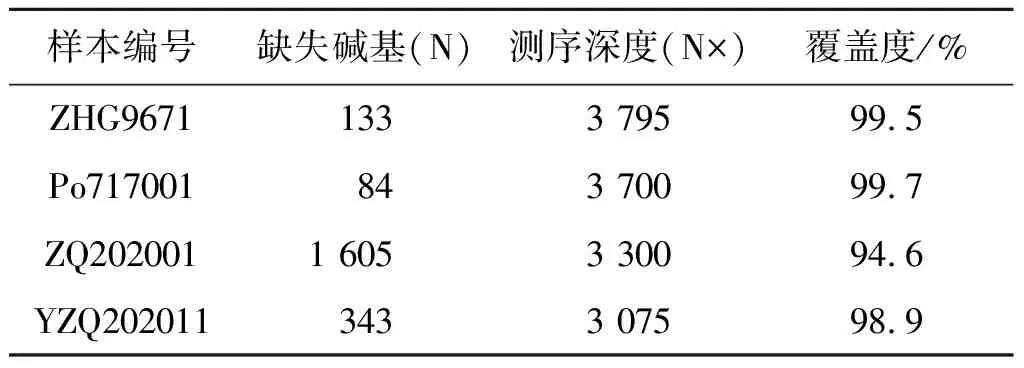
表2 基因組測序結果Tab.2 Results of the genome sequencing
2.2 核苷酸及氨基酸變異情況 以SARS-CoV-2 Wuhan-Hu-1株作為參考基因組,測序結果顯示4份樣本共檢測到46個堿基位點突變,其中涉及氨基酸性質(zhì)改變的非同義突變有27個,涉及5個編碼區(qū)域,其中導致ORF1編碼區(qū)氨基酸變異有12個、N基因編碼區(qū)6個、S基因編碼區(qū)6個、ORF3基因編碼區(qū)2個和ORF9b基因編碼區(qū)1個。除早期武漢居住史的病例ZQ202001,其余樣本在S基因編碼區(qū)均出現(xiàn)D614G的變異。位點變異情況詳見表3。

表3 位點變異情況Tab.3 Mutations of nucleotide and amino acid
2.3 系統(tǒng)進化樹分析 全基因組測序分析結果顯示,珠海市4份不同時期SARS-CoV-2病例來源于不同的進化分支,早期武漢輸入病例ZQ202001分屬于B,其核苷酸突變只與參考基因組SARS-CoV-2 Wuhan-Hu-1株相差1個堿基數(shù),而2020年7、8月鑒定的來自菲律賓的境外輸入病例ZHG9671與香港輸入病例Po717001同屬于B.1.1.63分支,與同時期香港公布的序列核苷酸相似性較高,遺傳距離相近。2020年12月鑒定的香港輸入病例YZQ202011分屬于B.1.36分支。見圖1。
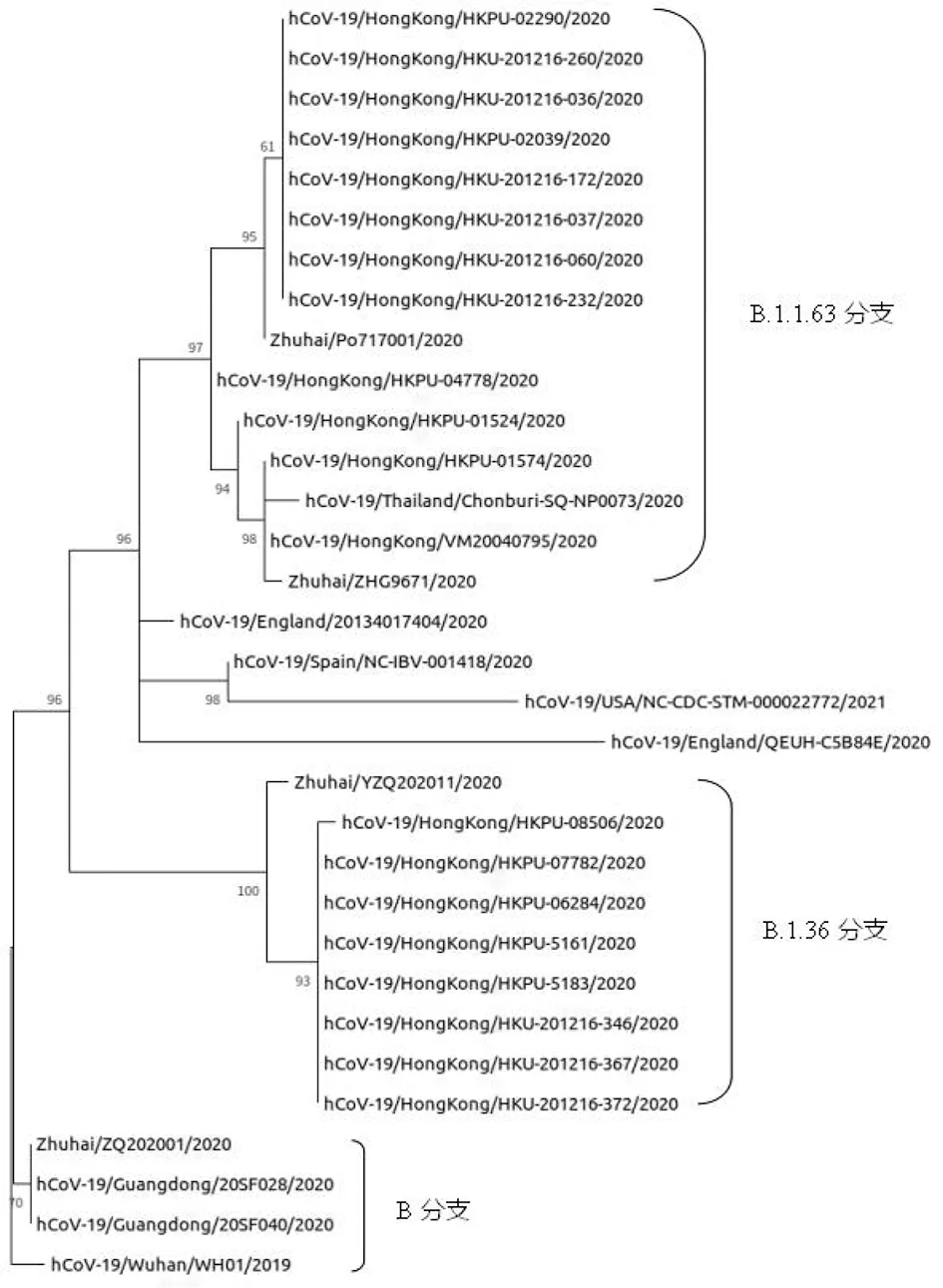
圖1 珠海市不同時期SARS-CoV-2全基因組系統(tǒng)發(fā)育進化圖Fig.1 Phylogenetic tree based on SARS-CoV-2 complete genomes in different decades in Zhuhai city
3 討 論
2020年1月底,SARS-CoV-2病毒刺突蛋白出現(xiàn)D614G突變,使得突變株陸續(xù)成為了后期的全球流行株[5]。目前,SARS-CoV-2經(jīng)過在全球不同人群間突變進化與新冠疫苗選擇壓力的雙重選擇下,已演變成800多種不同的亞型或分支[6],一些突變株如501Y.V2南非突變株、B.1.617印度流行變異株的出現(xiàn)導致了新一輪疫情的暴發(fā)。本次研究選取的主要為2020年不同時期COVID-19病例的標本,基因組測序結果顯示珠海市檢測的疫情早期樣本ZQ2020014與參考株SARS-CoV-2 Wuhan-Hu-1株的基因組高度相近,提示COVID-19大流行早期基因組保持著較低的進化水平,多數(shù)堿基突變導致氨基酸保守置換,對病毒的表型并未發(fā)生潛在的影響,與相關文獻報道結論一致[7]。其余3份樣本為全球疫情大流行期間檢出的境外輸入病例,共檢測到45個堿基位點突變,其中導致氨基酸性質(zhì)改變的突變有26個,以ORF1編碼區(qū)氨基酸突變頻率最高,不同于已報道的基因組變異均勻分布于各編碼區(qū)[8-9],推測可能是選取例數(shù)太少所導致的偏差。
2020年5月以后,國內(nèi)疫情逐漸趨于穩(wěn)定,珠海市的COVID-19病例以境外輸入為主,基因組測序分結果顯示,來自菲律賓的境外輸入病例ZHG9671與香港輸入病例Po717001同屬于B.1.1.63分支,2020年12月鑒定的香港輸入病例YZQ202011分屬于B.1.36分支,3份輸入病例標本均與同時期香港公布的序列核苷酸相似性較高,遺傳距離相近。從全基因組系統(tǒng)發(fā)育分析中可發(fā)現(xiàn),各個國家上傳的病毒基因組不僅在本國有聚集性的出現(xiàn),并且在多個分支中可發(fā)現(xiàn)有許多不同國家的散發(fā)株,這說明SARS-CoV-2在全球范圍內(nèi)穩(wěn)定持續(xù)傳播著,世界范圍內(nèi)的高密度人口流動可使病毒在短時間內(nèi)擴散,此外目前研究已報道世界范圍內(nèi)有大量的新冠感染者癥狀輕微或無癥狀[10],無癥狀感染者在COVID-19流行期間是潛在的強傳染源,具有隱匿性,臨床上不易被發(fā)現(xiàn)。亟需在目前篩查密切接觸者、高風險地區(qū)人員篩查以及感染源調(diào)查的基礎加強大數(shù)據(jù)的追蹤管理,加入定期核酸監(jiān)測來管控。
目前,對COVID-19臨床樣本的基因組測序多采用二代測序、三代測序平臺完成。第三代Nanopore納米孔測序技術屬于單分子測序,它根據(jù)將一個納米孔蛋白固定在電阻膜上,通過物理手段使DNA雙鏈解鏈成單鏈,利用馬達蛋白牽引DNA單鏈傳過納米孔,因不同堿基攜帶不同電荷在通過納米孔時引起電阻膜上電流的變化識別堿基,實現(xiàn)高通量測序。三代測序具有測序讀長長(超過150 kb)、速度快、測序數(shù)據(jù)實時監(jiān)控、機器方便攜帶等特點。Lu等[11]曾采用不同測序平臺對廣東省疫情早期的臨床病例樣本進行測序,結果顯示Nanopore混樣建庫PCR測序表現(xiàn)良好,尤其適用于病毒載量低的樣本。本次研究所選取的4份樣本均可獲得27 493~29 810 bp長度的基因組序列數(shù),基因組覆蓋度94.6%~99.7%不等,但樣本ZQ2020001和YZQ202011基因組序列缺失較多,測序質(zhì)量與病毒載量、文庫構建情況、原始標本核酸完整性密切相關,CT<30的臨床標本均能獲得較好的測序結果,本研究中的ZQ2020001和YZQ202011初次檢測CT值雖均在30以下,但基因組缺失較多,可能是在檢測過程中操作不當導致的核酸降解。對于COVID-19臨床標本,應在檢測陽性時及時提取全基因組進行疫源追蹤,核酸反復凍融對核酸的穩(wěn)定性及完整性均有較大影響,不利于后續(xù)基因組測序。
綜上所述,本次研究對本市COVID-19部分臨床標本進行全基因組測序,溯源分析顯示基因組之間核苷酸突變具有多態(tài)性,4份臨床標本均具有外地旅行史,均為輸入病例,未出現(xiàn)本地感染。三代測序技術可有效用于原始樣本中新型冠狀病毒基因組序列的溯源分析,在地市級疾控應用前景較好。
利益沖突:無
引用本文格式:龍冬玲,黃輝濤,魏泉德,等.珠海市不同時期新型冠狀病毒基因組測序及溯源分析[J].中國人獸共患病學報,2021,37(11):1003-1007. DOI:10.3969/j.issn.1002-2694.2021.00.138
