液相色譜-四極桿飛行時間質譜用于快速篩查動物組織中多種激素殘留的方法研究和應用
2021-12-16 05:39:14葉磊海葉佳明裘鈞陶鐘世歡詹越城王慶玲
食品工業科技 2021年24期
關鍵詞:方法
葉磊海,葉佳明,裘鈞陶,鐘世歡,黃 南,詹越城,何 斌,王慶玲
(1.浙江公正檢驗中心有限公司,浙江杭州 310009;2.贊宇科技集團股份有限公司,浙江杭州 310009)
激素是一類調節機體代謝,協調機體器官系統之間活動并維持內環境穩定的物質,主要包括糖皮質激素,性激素和孕激素[1]。近些年來,其快速高效的治療效果誘使人們不但將其用于動物疾病的治療,而且添加到動物飼料或飲用水中用于非治療性的防病和促生長,提高蛋白轉化率,改善畜產品品質,在動物養殖行業中非法使用[2],研究表明,某些激素可長期在動物體內中殘留,過量攝入后會對人體正常激素平衡造成干擾,引起體內分泌功能紊亂,抑制機體的免疫功能,誘發感染,甚至有致癌風險[3]。因此,歐盟、美國食品藥物管理局嚴格規定禁止在動物源性食品使用激素類藥物[4],我國農業部250號公告[5]也明確規定禁止在動物性食品中檢出甲基睪丸酮與群勃龍等激素類藥物。食品中獸藥最大殘留限量國家標準中也對部分藥物進行了限量規定,比如倍他米松在動物肌肉中限量 0.75 μg/kg,地塞米松 1.0 μg/kg[6]。
目前國內外檢測激素藥物的方法[7]主要有氣相色譜質譜法(GC-MS)[8]、高效液相色譜法(LC)[9]和液相色譜質譜串聯法(LC-MS)[10-11]。其中;GC-MS方法樣品需要衍生化,前處理繁瑣,有的激素也不容易通過衍生得到很好的極性和穩定性,制約了其在多殘留分析中的應用[12-14];LC由于檢測器的局限性,定性能力差,難以應用于多組分的的篩查和確證;LC-MS由于具有高靈敏和高選擇性的特點,被廣泛應用在獸藥,農殘的檢測工作。目前以四極桿飛行時間質譜(Q-TOF/MS)[15-16]等為代表的高分辨質譜技術為多組分藥物殘留的篩查帶來了新的手段,其具有高質量準確度、高質量分辨率,可提供全面的定性和定量信息等優點,通過精確質量數的自動檢索匹配以及二級特征碎片離子的確定,來實現復雜基質中痕量組分的高通量的篩查和確證。
QuEChERS作為一種常用的分散固相萃取技術被越來越多應用到高通量藥物篩查工作中,一些傳統的吸附劑如:十八烷基鍵合硅膠(C18),N-丙基乙二胺(PSA),石墨碳黑(GCB),中性氧化鋁(Alu-N),其中PSA和GCB能吸附基質中的糖類和色素,C18和Alu-N具有良好除脂能力,但是針對性較差,在去掉基質干擾物的同時也會吸附目標物。已有研究表明,一些新的材料憑借其某些特殊的性能被用來作為新型吸附劑用于農殘,抗生素和激素多殘留的檢測[17]。其中增強型脂質去除吸附劑(EMR)作為一種新型的吸附劑,采用特殊聚合物基質專門吸附脂質中C5及以上的碳鏈,對脂質具有非常強的選擇吸附性,官能化聚苯乙烯/二乙烯苯(PEP-2)吸附劑,其表面鍵合有吡咯烷酮和微量的脲基官能團,具有富于電荷的表面結構,可以吸附大多數高極性化合物。
測定激素藥物的前處理方法主要有液液萃取法[18]、固相萃取法[19]、QuEChERS(quick, easy,cheap, effective, rugged and safe)法[20]。液液萃取法要使用大量的有機試劑且步驟繁瑣,固相萃取法雖然凈化效果好,但使用的萃取小柱成本過高,QuEChERS早期應用于農藥殘留的檢測,隨著不同種類的新型吸附劑的開發,近年來也逐漸在測定激素工作中得到應用。本研究以常用32種激素為研究對象,旨在建立一種高效液相色譜一四極桿飛行時間質譜(LC-Q-TOF/MS)同時測定動物組織32種激素殘留的快速篩查和定量方法。方法擬用增強型脂質去除劑(EMR),N-丙基乙二胺(PSA)和官能化聚苯乙烯/二乙烯苯(PEP-2)等吸附劑代替傳統的吸附材料,以解決傳統QuEChERS所用的吸附劑在除脂和目標物共吸附的矛盾,以期為此類物質的檢測提供更多的技術選擇。
1 材料與方法
1.1 材料與儀器
32種標準物質 純度均大于95.0%,德國Dr.Ehrenstorfer,具體信息見表1;乙腈、甲酸、甲醇 色譜純,美國J.T.Baker;氯化鈉、硫酸鎂 均為分析純,國藥集團化學試劑有限公司;增強型脂質去除吸附劑(EMR) 美國 Agilent公司;十八烷基鍵合硅膠C18、N-丙基乙二胺(PSA)、石墨化碳黑(CCB)、中性氧化鋁(Al-N)、官能化聚苯乙烯/二乙烯苯(PEP-2)吸附劑 天津博納艾杰爾公司;實驗用水 Millipore-iQ高純水凈化儀制得;牛肉、雞肉、豬肝 均采自農貿市場,均質后-18 ℃保存待用。

表1 32種激素質譜篩查參數Table 1 Screening parameters of 32 hormones by mass spectrometry
Agilent 1290高效液相色譜-6545四極桿飛行時間質譜聯用儀 美國Agilent公司;SOP萬分之一電子分析天平 賽多利斯科學儀器有限公司;HS260旋渦振蕩器 德國IKA公司;N-EVAPl l 2水浴式氮吹儀 美國Organomation公司;Centrifuge 5804R高速冷凍離心機 德國Eppendorf公司。
1.2 實驗方法
1.2.1 樣品前處理 準確稱取5.0 g(精確到0.01 g)混合均勻的樣品于50 mL離心管中,加入5 mL水,旋渦混勻30 s,準確加入乙腈15 mL,超聲提取10 min,5000 r/min離心5 min,取上清液于另一離心管中,用乙酸乙酯10 mL重復提取殘渣1次,合并兩次上清液,加入1 g EMR吸附劑(預先加5 mL水進行活化),旋渦 30 s,然后 5000 r/min離心5 min,取全部上清液加入鹽析劑(3 g無水硫酸鎂和1 g氯化鈉),旋渦混勻30 s,然后5000 r/min離心5 min ,取全部上層清液加入100 mgPSA和10 mg官能化聚苯乙烯/二乙烯苯(PEP-2),旋渦混勻 30 s,然后 5000 r/min離心5 min,取上清液在40 ℃下用氮氣吹至近干,加入 1 mL 0.1% 甲酸水-乙腈(90:10,V:V)復溶,旋渦30 s,過 0.22 μm 濾膜上機。
1.2.2 色譜條件 色譜柱為Agilent Eclipse Plus-C18柱(3.0 mm×100 mm,1.8 μm),流動相:A:0.1% 甲酸,B:0.1% 甲酸乙腈,梯度洗脫程序:0~3 min,10%~30% B%,3~10 min,30%~75% B%,10~13 min,75%~75% B%,13~15 min,75%~90% B%,15~16 min,90%~90% B%,16~16.1 min,90%~10% B%,16.1~20 min,10%~10% B%。流速:0.3 mL/min ,溫度:30 ℃,進樣量:5 μL。
1.2.3 TOF/MS條件 離子源:Dual AJS ESI源,掃描方式:正離子全掃描采集一級質譜數據,Target MS/MS模式采集二級質譜數據;掃描范圍:m/z 50~700;毛細管電壓:4000 V;鞘氣溫度:250 ℃;鞘氣流速:12 L/min;干燥氣溫度:300 ℃;干燥氣流速:10 L/min;裂解電壓:150 V;飛行時間管真空度:2.05×10-7Torr;使用嘌呤(m/z 121.050873)和六磷氮(m/z 922.009798)作為參比液進行實時質量校準,質量誤差精度低于5×10-6。
1.2.4 數據庫的建立 用甲醇溶液分別配制各標準物質為100 μg/mL,并用50%甲醇水配制成質量濃度1.0 μg/mL 32種混合標液,為按上述儀器條件,在全掃描模式下對32種混合標液進行分析,獲得化合物的精確分子質量,同位素信息和保留時間,建立一級譜庫。然后在Target MS/MS模式下分析,得到不同碰撞能(10、20、40 eV)下碎片離子的質譜圖,建立二級譜庫,最后將兩次采集的信息導入PCDL軟件,完成質譜庫的建立。32種激素的分子式、精確質量數、質量偏差、保留時間、子離子等質譜參數見表1。
1.3 數據處理
數據分析采用安捷倫公司的質譜定性分析數據處理軟件(MassHunter Qualitative Analysis B.5.00)。樣品分別在全掃描模式和Target MS/MS模式進行數據采集,通過建立好的32 種激素類化合物數據庫進行化合物提取與匹配,主要通過保留時間、精確質量數、化合物同位素豐度進行定性。通過基質提取液配制標準曲線。空白樣品按步驟1.2.1方法進行處理,得到空白基質,以此配制質量濃度分別為5、10、20、50、100、200、1000 μg/L 的標準溶液,LC-QTOF/MS進樣測定,以各化合物的一級母離子響應峰面積對其質量濃度繪制曲線,外標法定量。
2 結果與分析
2.1 前處理條件的優化
2.1.1 提取劑的選擇 對目標物的提取效果取決于樣品的基質和化合物的極性,由于動物組織中基質復雜,含有蛋白質、脂肪等內源性物質,干擾目標物的分析,使得性激素多殘留分析檢測具有一定的難度[21-23],試驗涉及到的激素類藥物屬于弱極性或中等極性化合物,易溶于有機溶劑[24]。因此,本實驗選取雞肉、牛肉、豬肝作為不同類型的基質代表,以甲醇、乙腈、乙酸乙酯為提取劑考察不同提取劑的提取效果,具體見表2。結果表明,在50 μg/kg的加標水平下,三種提取劑甲醇提取效果較差,個別組分如丙酸諾龍單次提取效率只有20%~40%,乙腈和乙酸乙酯單次提取效率較高,但是在豬肝復雜基質中個別組分如孕酮,在乙腈中的提取效率要高于乙酸乙酯,而在雞肉等簡單基質中,孕酮在乙酸乙酯中的提取效率要好于乙腈。可能是由于在復雜基質中乙酸乙酯提取共萃物較多,導致更多的基質效應。因此本實驗采用乙腈和乙酸乙酯分步提取的方式,能夠兼顧不同基質不同組分的提取效率,兩次提取后各藥物平均回收率在85%以上。

表2 32種激素中不同基質中3種提取劑回收率的比較Table 2 Comparison of recovery rates of three extractants in different substrates of 32 hormones
2.1.2 凈化條件的選擇 本研究通過加標回收率考察了 C18、Alu-N、GCB、EMR、PSA、PEP-2這 6種吸附劑對32種激素的吸附情況。結果表明C18、Alu-N、GCB對目標化合物均有不同程度的吸附,其中C18、Alu-N對丙酸睪酮、氫化可的松等親脂性較強的激素有明顯吸附,平均回收率在80%左右,GCB因其可吸附帶苯環官能團的藥物,對大部分藥物均有明顯的吸附,平均回收率不足70%。而適量的EMR、PSA和PEP-2對化合物的吸附影響較小,回收率在90%以上,具體見圖1。通過平均回收率和基質效應結果,進一步考察了不同添加量的凈化效果,參考文獻[25]和經驗,分別比較了在100 μg/L的混合標準溶液中添加 EMR 1~5 g,PSA 100~500 mg,PEP-2 10~50 mg不同添加量的凈化效果,具體見表3,結果表明:EMR不同添加量回收率和基質效應基本沒有差別,PSA加入100 mg時,基質效應適中,回收率最佳,而PEP-2在加入量超過30 mg的時候,平均回收率不到75%,加入量在減少至10 mg,平均回收率在90%以上,與不加入對比,樣液的澄清度和圖譜上雜質峰情況有明顯改善。所以本實驗最終確定加入1 g EMR吸附劑,100 mg PSA,10 mg PEP-2作為最終的添加量。

表3 3種吸附劑不同加入量對32種目標物回收率的影響Table 3 Efects of different amounts of three adsorbents on the recoveries of 32 target compounds

圖1 不同吸附劑對32種目標物回收率的影響Fig.1 Effect of different adsorbents on the recovery of 32 target compounds
吸附劑實驗表明,基于增強型脂質吸附劑(EMR)的QuEChERS法能夠解決傳統吸附劑與目標物共吸附的矛盾,可以去除基質中大部分雜質,很大程度上降低了基質干擾,提高整個方法的靈敏度。
2.2 質譜和色譜條件的優化及數據庫的建立
2.2.1 質譜條件優化和數據庫的建立 使用Dual AJS ESI源,在m/z 50~700范圍內對32種1.0 μg/mL目標化合物進行一級質譜全掃描分析,得到目標化合物的總離子色譜圖,結果顯示,所有藥物在正離子得到的碎片信息要多于負離子模式,正離子模式下形成[M+H]+準離子峰。比較各個化合物提取離子流色譜圖的響應和峰型,依次對離子源參數(毛細管電壓、干燥器溫度、干燥器流速、鞘氣流速、鞘氣溫度等)進行優化,使質譜儀靈敏度達到最優,然后優化各化合物的裂解電壓,使其響應值最大,其范圍在120~170V,最終選擇均能獲得較高響應值的150 V作為本方法的裂解電壓。
飛行時間質譜主要利用各化合物的篩查參數來篩查和確證目標物,本方法主要通過Agilent Mass-Hunter Qualitative Analysis(B.5.00)軟件完成化合物質譜篩查參數數據庫的建立。在全掃描模式下先進行一級質譜掃描,得到目標化合物的保留時間、精確分子質量、同位素信息、質量誤差等相關信息,然后在Target MS/MS模式下采集得到碰撞能為10、20、40 eV各化合物的二級質譜數據,建立32種目標化合物的篩查數據庫。根據歐盟2002/657/EC法案的中累計4個識別點鑒定化合物的要求,高分辨質譜母離子得到2個識別點,碎片離子得到2.5識別點。因此基于保留時間,一級母離子飛行時間質譜精確質量數、同位素豐度匹配、二級特征離子碎片等信息的飛行時間高分辨質譜具有比低分辨質譜更可靠的定性分析優勢。
2.2.2 色譜條件優化 32種激素在C18色譜柱上均有較好的保留,考慮到選擇的激素藥物中有多種同分異構體,主要有潑尼松龍和可的松,地塞米松和倍他米松,曲安奈德、倍他米松醋酸酯、地塞米松醋酸酯和氟尼縮松,阿氯米松雙丙酸酯和倍氯米松雙丙酸酯,甲基潑尼松龍醋酸酯和地索奈德共5組同分異構體,給分離造成了一定的困難,根據經驗和文獻[26-27],主要通過不同的色譜柱和不同的流動相及梯度洗脫體系進行了優化。本研究主要比較Extend C18、BEH C18、Eclipse Plus-C18三種色譜柱對32種激素的保留行為,結果表明目標化合物包括5組同分異構體在EclipsePlus-C18色譜柱能獲得較理想的分離度。
同時,對比了甲醇水、乙腈水體系兩種流動相體系,結果發現,采用甲醇-水流動相時,幾種同分異構體無法有效分離,而乙腈-水體系不僅能有效分離同分異構體,而且峰型更尖銳,響應值更高,主要是因為乙腈較甲醇有更強的洗脫能力。所以,選擇乙腈為有機流動相,再進一步比較不同pH的水系流動相(水、0.1%甲酸、0.2%甲酸、1%甲酸水溶液)。結果表明,在水中加入0.1%甲酸,能提高目標化合物的響應,并獲得滿意的分離效果,為了保持流動相體系的平衡,在乙腈中也加入0.1%甲酸。故最終選擇0.1%乙腈-0.1%甲酸水作為本方法的流動相體系,EclipsePlus-C18色譜柱作為分離工具,可以兼顧32種激素藥物的色譜保留行為及質譜響應,經優化后的梯度洗脫能在20 min內有效分離32種化合物。在以上條件優化后的離子色譜圖2。

圖2 32種激素提取離子色譜圖Fig.2 High resolution extracted ion chromatogram of 32 hormones
2.3 基質效應的研究
基質效應是利用液相色譜-質譜聯用分析復雜藥物時影響定量準確性的重要因素[28],共流出在離子源競爭性離子化,會導致目標物離子化效果的增強或抑制,從而表現出基質增強或者抑制現象[29-30]。本實驗以豬肉為研究對象,在空白豬肉提取液中加入不同濃度的混合標液,并與空白甲醇試劑配制的標液進行比較,以Matrix Effect(ME)表示基質效應,本實驗每種目標物的基質效應結果如表4所示,ME值越接近1,說明基質效應越小,反之亦然。結果表明,在優化后方法處理后,各藥物的基質效應在0.725~0.913,具體見表4,說明本方法的前處理能有效降低基質效應,為了獲得更準確的定量結果,本方法還是采用基質標校準曲線進行定量計算。本方法也可以通過使用內標物降低基質效應影響,但是考慮到內標物造價昂貴,很難找到滿足所有藥物篩查條件的內標物,因此本實驗并未采用添加內標的方法。
2.4 方法學考察
2.4.1 線性范圍、回歸方程、檢出限和定量限 在本文確定條件下,對32種激素藥物進行測定,用牛肉空白基質配制一系列濃度,以質量濃度為橫坐標,峰面積為縱坐標建立標準曲線,采用標準加入法進行測定,以一級母離子響應3倍信噪比和10倍信噪比分別確定樣品的檢出限(LOD)和定量限(LOQ),通過表4可以看到32種藥物在各自線性范圍內相關系數≥0.995以上,線性相關良好,LOD范圍為1~10 μg/kg,LOQ范圍為 3~30 μg/kg,靈敏度滿足實際檢測的需要。

表4 32種激素的基質效應、回歸方程、相關系數、線性范圍、檢出限(LOD)及定量限(LOQ)Table 4 Matrix effect, regression equation, correlation coefficient, linear range, LOD and LOQ of 32 hormones
2.4.2 方法回收率和精密度驗證 選取牛肉、雞肉、豬肝不同基質陰性樣品分別添加10、50、100 μg/kg 3個不同濃度,每個濃度進行6次重復測定,考察方法回收率和精密度,從結果來看,該方法回收率在不同基質中達到70.3%~116.2%之間,相對標準偏差(RSD)為0.87%~8.97%,說明本方法具有良好的精密度和準確度。其中牛肉中加標回收率和RSD具體見表5,其他數據不一一列出。

表5 牛肉中32種激素加標回收率和相對標準偏差(n=6)Table 5 Recoveries and relative standard deviations of 32 hormones in beef (n=6)
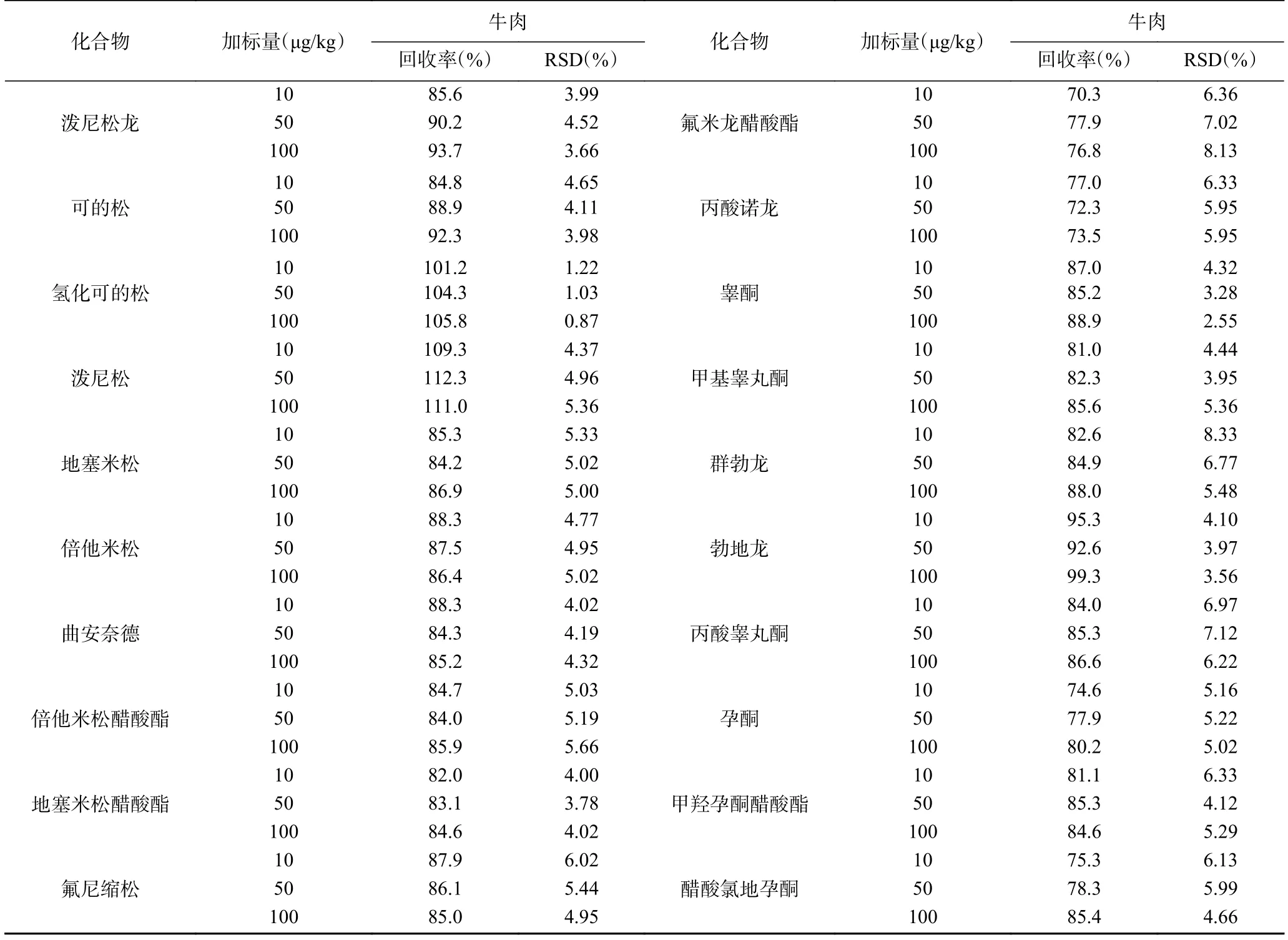
續表5
2.5 實際樣品的篩查
采用本方法對市售30批次雞肉,30批次牛肉和20批次豬肝進行檢測,每種樣品進行雙平行測定,其中檢測出豬肝中地塞米松1份,雞肉中地塞米松 1份,含量分別為 10.2、6.7 μg/kg,均超過最大殘留限量1.0 μg/kg規定[6],陽性樣品用國標方法進行方法驗證,測量結果非常接近。表明本方法結果可靠,可作為日常樣品的篩查。
3 結論
本方法建立了基于QTOF高分辨質譜快速篩查動物組織中多種激素殘留檢測方法,通過乙腈和乙酸乙酯分步提取,提取劑用EMR、PSA、PEP-2分步凈化,通過C18色譜柱進行分離,在全掃描模式下采集一級質譜數據,以待測物的準分子離子峰的峰面積定量,以保留時間、精確質量數、同位素豐度比等特征信息定性,并應于于市售80份實際樣品的篩查分析。結果說明,該方法操作簡單,回收率高,重現性好。80份樣品檢測出豬肝中地塞米松 1 份,雞肉中地塞米松 1 份,含量分別為 10.2、6.7 μg/kg,均超過最大殘留限量 1.0 μg/kg 規定。采用一次制備樣品,可以同時快速篩查動物組織中32種激素藥物,一次進樣分析,可同時提供定量和定性的結果,大大提高實驗室檢測能力,為進一步準確篩查動物組織中激素殘留提供了豐富的技術手段。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
