溶酶體組織蛋白酶B增加自噬保護缺氧誘導的心臟微血管內皮細胞損傷
2022-01-14 09:22:52李亞彭肖莉麗張彥周
中國藥理學通報 2022年1期
關鍵詞:小鼠
高 路,姚 瑞,李亞彭,梁 翠,肖莉麗,張彥周
(鄭州大學第一附屬醫院心血管內科,河南 鄭州 450052)
心血管病現患人數3.30億,其中冠心病1 100萬,心力衰竭890萬[1]。隨著心肌梗死患者接受血管再通治療的比例增多,心肌梗死后存活患者比例增高,這部分患者最終會發展成為心力衰竭[2]。心肌梗死中缺氧導致血管內皮細胞損傷是心肌梗死后心力衰竭病理發展的主要原因[3]。在缺氧刺激下,內皮細胞發生炎癥、氧化應激和凋亡,導致內皮細胞增殖減少、舒張功能障礙、細胞數量減少[4]。此外,內皮細胞自噬流受阻是缺氧誘導內皮細胞損傷的主要病理改變[5]。因此,明確缺氧條件下內皮細胞損傷的機制,對發現防治心臟缺血損傷新的干預靶點至關重要。
溶酶體組織蛋白酶B(CTSB)是一種溶酶體酶,其在維持溶酶體功能、細胞凋亡和自噬中發揮重要作用[6]。研究表明CTSB在心血管疾病中發揮重要作用。Mehra等[7]報道CTSB在擴張型心肌病患者血漿中表達增高與射血分數降低密切相關。CTSB可通過激活炎癥小體和促進細胞凋亡而加劇柯薩奇病毒B3誘導的心肌炎[8]。以往的研究發現,CTSB基因敲除小鼠可通過TNF-α-ASK1-JNK途徑減輕壓力負荷下的心肌重構[9]。但是CTSB是否可以通過調控自噬參與缺氧誘導的內皮細胞損傷尚不清楚。本研究擬通過分離培養CTSB基因敲除小鼠心臟微血管內皮細胞,探討CTSB在缺氧誘導的內皮細胞損傷中的作用。
1 材料與方法
1.1 材料CTSB基因敲除(KO)小鼠購買于Jackson Lab(貨號: 030971,美國);C57BL6J野生型(WT)小鼠購買于北京華富康公司;TNF-α、IL-1、IL-6 ELISA試劑盒購買于Biolegend公司(貨號:430901, 432604, 431304美國);CD31(貨號:ab28364)、鈣粘蛋白(Cadherin,貨號:ab33168)一抗購買于Abcam公司(美國);LC3-GFP-mCherry雙標腺病毒購買于山東維真公司;Tunel染色試劑盒購買于Millipore公司(貨號:S7111,美國);巴弗洛霉素(BAF,貨號:19-148)購買于Sigma公司;caspase-3活性檢測試劑盒購買于碧云天公司(上海,貨號:C1115);CTSB過表達腺病毒購買于山東維真公司。
1.2 方法
1.2.1內皮細胞分離培養 4~6周C57BL6J小鼠或CTSB基因敲除小鼠心臟剪出后置于D-Hanks平衡液中沖洗,心臟剪碎后采用膠原酶在37 ℃消化心臟15 min,分5次消化后收集消化液,置于終濃度為10%FBS的DMEM-F12中,將細胞過濾后重懸,采用CD31磁珠吸附內皮細胞,將內皮細胞置于含10%FBS的DMEM-F12中培養。
1.2.2內皮細胞分組處理
1.2.2.1 第一部分 實驗分組:① WT-常氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用常氧(5%CO2,95%空氣)培養48 h;② KO-常氧組:分離CTSB基因敲除小鼠心臟微血管內皮細胞,采用常氧處理48 h。③ WT-缺氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用缺氧(5%氧氣,95%N2)培養48 h;④ KO-缺氧組:分離CTSB基因敲除小鼠心臟微血管內皮細胞,采用缺氧處理48 h。
1.2.2.2 第二部分 將細胞分組:① Ad-NC-常氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒空載體感染內皮細胞8 h,采用常氧培養48 h;② Ad-CTSB-常氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒感染內皮細胞過表達CTSB(Ad-CTSB)8 h,采用常氧處理48 h。
③ Ad-NC-缺氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒空載體感染內皮細胞8 h,采用缺氧刺激48 h;④ Ad-CTSB-缺氧組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒感染內皮細胞過表達CTSB(Ad-CTSB)8 h,缺氧處理48 h;⑤ Ad-NC-常氧+巴弗洛霉素組(BAF)組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒空載體感染內皮細胞8 h,BAF (100 nmol·L-1)處理12 h,常氧處理48 h;⑥ Ad-CTSB-缺氧+BAF組:分離C57BL6J野生型小鼠心臟微血管內皮細胞,采用腺病毒感染內皮細胞過表達CTSB 8 h,BAF(100 nmol·L-1)處理12 h,缺氧處理48 h。
1.2.3內皮細胞CD31和鈣黏連蛋白免疫熒光染色 采用CD31和鈣黏連蛋白免疫熒光染色鑒定分離的心臟微血管內皮細胞。細胞采用4%多聚甲醛固定5 min,采用0.1%TritonX通透后,采用8%羊血清封閉,采用抗CD31抗體和抗鈣黏連蛋白抗體(1 ∶100稀釋)4 ℃孵育過夜,采用Alexa FluorH 488羊抗小鼠和568羊抗兔二抗孵育1 h,采用DAPI進行細胞核染色,熒光顯微鏡下觀察。
1.2.4ELISA檢測炎癥細胞因子的釋放 各組處理完畢后收集細胞,將細胞在含有RIPA的裂解液中裂解后在冰上放置10 min,根據Elisa試劑盒配置標準蛋白,將樣本和標準品加入酶標板中,在37 ℃孵育30 min,洗滌后加入100 μL酶結合物,洗滌后加入1μLTMD,15 min后加入硫酸終止液,在酶標儀(Synergy HT, BioTek, 美國)450 nm處讀取吸光度。根據標準品濃度制作標準曲線,計算各樣本的濃度。
1.2.5TUNEL染色檢測細胞凋亡 細胞采用4%多聚甲醛固定5 min,采用0.1%TritonX通透后,采用Tunel試劑酶在37 ℃孵育1 h,采用DAPI進行細胞核染色,熒光顯微鏡(OLYMPUS BX43,日本)下觀察。細胞凋亡數量計算單個視野內TUNEL陽性細胞數*100%/總細胞數。
1.2.6caspase-3活性檢測 使用RIPA裂解液裂解細胞,加入檢測緩沖液,再加待測樣品,隨后再加入10 μL Ac-DEVD-pNA(2mM),37 ℃孵育60 min。在酶標儀(Synergy HT,BioTek,美國)下檢測405 nm處吸光度。
1.2.7LC3-GFP-mcherry雙標病毒檢測細胞自噬流 細胞接種于24孔板,在處理前感染LC3-GFP-mcherry雙標病毒8 h,隨后進行各組處理,將細胞爬片取出,在熒光顯微鏡OLYMPUS BX43,日本)下計數細胞內紅色和綠色LC3數量。
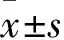
2 結果
2.1 CD31和鈣黏連蛋白染色鑒定內皮細胞采用CD31和鈣黏連蛋白免疫熒光雙染色鑒定分離培養的內皮細胞,結果顯示分離出的細胞均表達CD31和鈣黏連蛋白(Fig 1A),提示心臟微血管內皮細胞分離成功。采用免疫印跡鑒定內皮細胞CTSB蛋白的表達,結果顯示,KO組小鼠內皮細胞CTSB完全不表達,WT組內皮細胞正常表達CTSB蛋白(Fig 1B)。
2.2 CTSB敲除加重缺氧誘導的內皮細胞炎癥采用Elisa檢測野生型小鼠內皮細胞和CTSB基因敲除內皮細胞缺氧刺激后TNF-α、IL-1和IL-6的釋放,結果顯示:與WT-常氧組相比,WT-缺氧組內皮細胞TNF-α、IL-1和IL-6的釋放明顯增加(P<0.05);而KO-缺氧組內皮細胞TNF-α、IL-1和IL-6的釋放高于缺氧組(P<0.05),KO-常氧組與WT-常氧組之間上述炎癥因子的釋放無差異(P>0.05)。見Tab 1。
2.3 CTSB敲除加重缺氧誘導的內皮細胞凋亡采用TUNEL染色檢測細胞凋亡,結果顯示:與WT-常氧組相比,WT-缺氧組內皮細胞凋亡細胞數量明顯增加(P<0.05);而KO-缺氧組內皮細胞凋亡細胞數量明顯高于缺氧組(P<0.05);KO-常氧組與WT-常氧組細胞凋亡數量無差異(P>0.05),見Fig 2,Tab 2。

Fig 1 Identification of endothelial cells(×200)

Tab 1 Release of inflammatory factors in endothelial cells in each group (n=6)

Fig 2 Endothelial cell apoptosis aggravated by CTSB knockout (×200)

Tab 2 Endothelial cell apoptosis aggravated by CTSB knockout (n=6)

Fig 3 Autophagy flow in endothelial cells blocked by CTSB knockout (×400) Legend: Green represents GFP, and red represents mCherry
采用caspase-3活性檢測試劑盒檢測細胞凋亡執行器caspase-3的活性,結果顯示:與WT-常氧組相比,WT-缺氧組內皮細胞caspase-3活性明顯增加(P<0.05);而KO-缺氧組內皮細胞caspase-3活性明顯高于缺氧組(P<0.05);KO-常氧組與WT-常氧組caspase-3活性無差異(P>0.05),見Tab 2。
2.4 CTSB敲除阻斷內皮細胞自噬流采用LC3-GFP-mCherry雙標腺病毒檢測內皮細胞自噬流,結果顯示:與WT-常氧組比,WT-缺氧組LC3-GFP(綠色熒光點)數量明顯增多(P<0.05);而KO-缺氧組內皮細胞LC3-GFP數量多于WT-缺氧組(P<0.05)。KO常氧組LC3-GFP數量與WT-常氧組無差異(P>0.05)(Fig 3,Tab 2)。4組LC3-mCherry(紅色熒光)數量無差異(P>0.05),提示CTSB基因敲除導致自噬降解受阻。
2.5 內皮細胞腺病毒載體過表達CTSB第二部分實驗,采用腺病毒載體感染C57BL6J野生型小鼠來源的心臟微血管內皮細胞,免疫印跡結果顯示:當采用MOI=50時,Ad-CTSB感染效率最佳(Fig 4)。
2.6 CTSB過表達減輕缺氧誘導的內皮細胞炎癥采用ELISA檢測各組細胞TNF-α、IL-1和IL-6的釋放,結果顯示:與Ad-NC-常氧組相比,Ad-NC-缺氧組內皮細胞TNF-α、IL-1和IL-6的釋放明顯增加(P<0.05);而Ad-CTSB-缺氧組內皮細胞TNF-α、IL-1和IL-6的釋放低于缺氧組(P<0.05);Ad-NC-常氧+BAF組細胞炎癥因子的釋放高于Ad-NC-常氧組(P<0.05);Ad-CTSB-缺氧+BAF組高于Ad-CTSB-缺氧組(P<0.05),見Tab 3。

Tab 3 Release of inflammatory factors in endothelial cells in each group (n=6)

Fig 4 Endothelial cell adenovirus vector overexpression CTSB (n=6)
2.7 CTSB過表達減輕缺氧誘導的內皮細胞凋亡采用Tunel檢測細胞凋亡水平,結果顯示:與Ad-NC-常氧組相比,Ad-NC-缺氧組內皮細胞凋亡細胞數量明顯增加(P<0.05);而Ad-CTSB-缺氧組內皮細胞凋亡低于缺氧組(P<0.05)。Ad-NC-常氧+BAF組細胞細胞凋亡數量高于Ad-NC-常氧組(P<0.05);Ad-CTSB-缺氧+BAF組細胞凋亡數量高于Ad-CTSB-缺氧組(P<0.05),見Fig 5,Tab 4。
2.8 CTSB過表達改善內皮細胞自噬流采用LC3-GFP-mCherry雙標腺病毒檢測內皮細胞自噬流,結果顯示:與Ad-NC-常氧組相比,Ad-NC-缺氧組LC3-GFP數量明顯增多(P<0.05);而Ad-CTSB-缺氧組內皮細胞LC3-GFP數量低于缺氧組(P<0.05)。Ad-NC-常氧+BAF組LC3-GFP數量高于Ad-NC-常氧組(P<0.05);Ad-CTSB-缺氧+BAF組LC3-GFP數量高于Ad-CTSB-缺氧組(P<0.05)(Fig 6,Tab 4)。LC3-mCherry數量在6組之間無顯著性差異(P>0.05)。

Tab 4 CTSB overexpression reduces hypoxia-induced endothelial cell apoptosis and improves autophagy (n=6)
3 討論
內皮細胞排列在心血管系統的血管內,在維持心臟所需的氧氣和營養供應方面起著重要作用[10]。正常生理條件下,成人內皮細胞在血管內保持靜止,但是當受到外界刺激損傷時可迅速激活[11]。在缺氧條件下,過度的內皮細胞損傷可導致其本身舒張功能障礙,增殖減少,小管形成功能降低,內皮細胞凋亡增多。在心臟持續缺氧條件下,上述病理改變可導致心臟組織微血管數量減少,心肌細胞供氧減少,導致心肌細胞功能障礙和死亡,從而誘導心衰的發生發展[12]。因此,維持正常的心臟微血管內皮細胞功能對保護病理條件下心臟功能至關重要。我們的研究發現CTSB可參與缺氧條件下內皮細胞的損傷發生,在內皮細胞中缺失CTSB,可加重缺氧條件下內皮細胞的炎癥和凋亡;然而在內皮細胞中過表達CTSB可減輕缺氧條件下內皮細胞的炎癥和凋亡。細胞自噬在內皮細胞病理損傷中發揮重要作用。細胞自噬是一個保守的過程,它是細胞器和長壽蛋白通過囊泡和溶酶體系統降解的主要途徑。自噬水平的改變對細胞而言可能是保護性的,也可能是有害性的,這取決于自噬的程度和細胞環境[13-14]。Martello等[15]報道,內皮細胞基礎水平自噬降低可導致內皮細胞功能受損,心臟微血管密度降低。而Tang等[16]報道,通過減少自噬/線粒體自噬可保護內皮細胞缺氧/復氧損傷。目前對自噬在內皮細胞中的作用報道不一。主要原因在于多數研究并未檢測細胞自噬流的水平。作為自噬溶酶體降解的主要酶之一,CTSB在自噬溶酶體降解中發揮關鍵作用[17]。我們的研究采用CTSB全基因敲除小鼠,分離其心臟微血管內皮細胞。采用LC3-GFP-mcherry自噬雙標病毒標記自噬流的動態。在未發生自噬的細胞及含有自噬體的細胞中,由于mCherry與GFP共同表達,細胞呈現黃色熒光。當自噬體與溶酶體融合形成自噬溶酶體后,酸性的溶酶體環境使酸敏感的GFP熒光淬滅,而mCherry不受影響,進而使自噬溶酶體呈現紅色熒光[18]。因此,綠色熒光越多,提示自噬降解受阻。我們發現在缺氧條件下,GFP綠色熒光明顯增多,而紅色熒光不變,提示內皮細胞自噬降解明顯受阻。CTSB基因敲除內皮細胞發現在缺氧條件下,其綠色熒光明顯增加,紅色熒光與缺氧組無差異,提示CTSB基因敲除阻斷了內皮細胞自噬溶酶體的降解,細胞自噬流受阻,凋亡增多。

Fig 5 Endothelial cell apoptosis induced by hypoxia reduced by CTSB overexpression (×200)

Fig 6 Endothelial cell autophagy flow improved by CTSB overexpression (×400)
巴弗洛霉素是V-ATPase酶抑制劑,可抑制細胞質膜和溶酶體膜表面的V-ATPase酶,減少細胞質內H+離子向溶酶體轉運。巴弗洛霉素通過降低溶酶體的酸性環境減少自噬溶酶體的降解。本研究通過CTSB腺病毒載體感染內皮細胞,增加內皮細胞內CTSB蛋白的表達,發現細胞過表達CTSB能夠減輕缺氧誘導的炎癥和細胞凋亡。此外,CTSB蛋白過表達可增加自噬降解(表現為LC3-GFP綠色熒光明顯減少,而紅色熒光不變)。當采用巴弗洛霉素處理細胞后,CTSB蛋白過表達對內皮細胞的保護作用不再發生(表現為細胞炎癥和凋亡明顯增高)。提示CTSB通過調控自噬溶酶體降解在內皮缺氧損傷中發揮保護作用,CTSB保持自噬流的通暢是其保護內皮細胞缺氧損傷的基礎。
以往的研究報道,CTSB在擴張型心肌病中表達增高,其與射血分數降低密切相關。CTSB可加劇柯薩奇病毒B3誘導的心肌炎[8]。CTSB基因敲除小鼠可減輕壓力負荷下的心肌重構[9]。而這些研究均與我們的研究結果相悖。上述研究均在著重于CTSB在心肌細胞中的作用和機制,然而作為溶酶體蛋白之一,CTSB廣泛表達于除心肌細胞以外的其他細胞,如成纖維細胞、內皮細胞、免疫細胞等。其在不同的細胞中可能發揮不同作用。另外一種可能是CTSB單獨缺失,內皮細胞可能通過其他補償機制平衡自噬和凋亡(我們的結果發現單獨過表達或者敲除CTSB并不會影響內皮細胞自噬和凋亡),但是在合并缺氧的條件下,CTSB缺失誘發的自噬抑制能夠大大增加內皮細胞死亡。
綜上所述,CTSB參與內皮細胞缺氧損傷,CTSB通過增加自噬降解改善缺氧導致的內皮細胞自噬流阻塞,以CTSB作為靶點的治療方法可能成為延緩心肌梗死后心力衰竭的新手段。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
