用電解錳陽極液制備電池級硫酸錳
2022-02-18 08:03:08狄家康王啟任
濕法冶金 2022年1期
關鍵詞:質量
狄家康,王啟任,王 帥,鐘 宏,張 超
(1.中南大學 化學化工學院 錳資源高效清潔利用湖南省重點實驗室,湖南 長沙 410083;2.貴州武陵錳業有限公司,貴州 松桃 554115)
電解錳過程中,陽極液中會富集大量鎂離子。鎂離子濃度升高會導致晶體析出堵塞孔路、電解效果下降、金屬錳產品質量下降等問題[1]。因此,從電解錳陽極液中去除鎂離子是電解錳過程面臨的一個重要問題[2-6]。
近年來,鋰離子電池需求量逐年增大[7-8],而電池級硫酸錳是各類錳系鋰離子電池正極材料的基礎材料,其產量和質量直接影響電池質量[9-11]。
目前,工業上從溶液中去除鎂離子主要采用氟離子沉淀法[2,9-10],但該法對設備腐蝕性較大,對環境危害較大。采用溶劑萃取法分離錳、鎂效果較好、且易于連續操作[12-15],常用萃取劑有P204、P507[15-19]。試驗研究了用一種新型萃取劑A從模擬電解錳陽極液中選擇性萃取錳離子,并制備電池級硫酸錳,以期為高純硫酸錳溶液的獲得提供一種新萃取劑。
1 試驗部分
1.1 試驗原料與儀器
試劑:萃取劑A,磺化煤油,長沙晶康新材料科技有限公司;一水合硫酸錳、硫酸銨、氫氧化鈉,國藥集團化學試劑有限公司;無水硫酸鎂,碳酸錳,天津市科密歐化學試劑有限公司;硫酸,成都市科隆化學品有限公司。所有試劑均為分析純。水,去離子水。
模擬電解錳陽極液:Mn2+、Mg2+質量濃度分別為16.42、22.13 g/L。
試驗儀器:SHA-C恒溫振蕩器,DF-101S集熱式恒溫加熱磁力攪拌器,pH計(PHS-3C),電感耦合等離子體發射光譜儀(ICP-OES),電感耦合等離子體質譜儀(ICP-MASS)。
1.2 試驗原理與方法
萃取劑A為羧酸類萃取劑,屬于陽離子交換萃取劑,在用于萃取前需要皂化,皂化反應為:
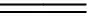
(1)
皂化萃取劑對溶液中金屬離子的萃取、反萃取反應可以簡單表示為:
萃取,

(2)
反萃取,
(3)
式中:Mn+—金屬離子;HA—酸性陽離子交換型萃取劑。
陽離子交換型萃取劑在實際萃取過程中的反應比式(2)要復雜得多,具體萃取過程這里不做討論。由反應式(2)、(3)看出,萃取劑A在低酸度環境下發生萃取反應,在高酸度環境下發生反萃取反應[20-22],這表示該萃取劑可循環使用。
有機相為萃取劑A-磺化煤油溶液。有機相中加入一定體積氫氧化鈉溶液(30%),放入恒溫振蕩器中皂化15 min,皂化結束后靜置分相,去除下層水相,得到一定鈉皂化有機相。萃取時,將皂化有機相與模擬電解錳陽極液按一定體積比加入帶塞錐形瓶中,放入恒溫振蕩器中振蕩一定時間,溫度25 ℃。振蕩萃取完成后,于分液漏斗中靜置分相。取初始水相和萃余液測定錳、鎂離子質量濃度,計算萃取率。水相中的錳、鎂離子質量濃度經稀釋后用ICP-OES測定。
負載錳的有機相和一定濃度硫酸溶液按一定體積比倒入帶塞錐形瓶中,放入恒溫振蕩器中振蕩反萃取15 min后取出,倒入分液漏斗靜置分相。取反萃取液測定錳和余酸質量濃度,計算反萃取率。
反萃取液經活性炭吸附去除有機相,用高純碳酸錳溶液調pH,之后抽濾,濾液加入到蒸發器中緩慢升溫蒸發、濃縮結晶得到高純硫酸錳晶體,晶體干燥后得到一水合硫酸錳產品。產品中的雜質以ICP-MASS法測定。
2 試驗結果與討論
2.1 溶劑萃取
2.1.1 溶液pH對錳、鎂萃取率的影響
用高濃度硫酸溶液和氫氧化鈉溶液調溶液pH為1~6,萃取相比Vo/Va=1.5/1,萃取劑體積分數30%,有機相皂化率30%,溫度25 ℃,萃取時間15 min,水相pH對錳、鎂萃取率的影響試驗結果如圖1所示。可以看出:水相初始pH較低時,溶液中存在大量氫離子,抑制了萃取劑和金屬離子的結合,錳、鎂離子萃取率均非常低;隨pH升高,錳萃取率快速升高并在pH=3左右時達最高,為75%左右,而鎂萃取率變化不大,維持在15%左右;此時,錳、鎂分離系數為18.33;而pH升至6時,錳萃取率略有下降,這可能是錳離子在高pH條件下不穩定所致。因溶液初始pH為4.6,所以,試驗過程中不必調節。
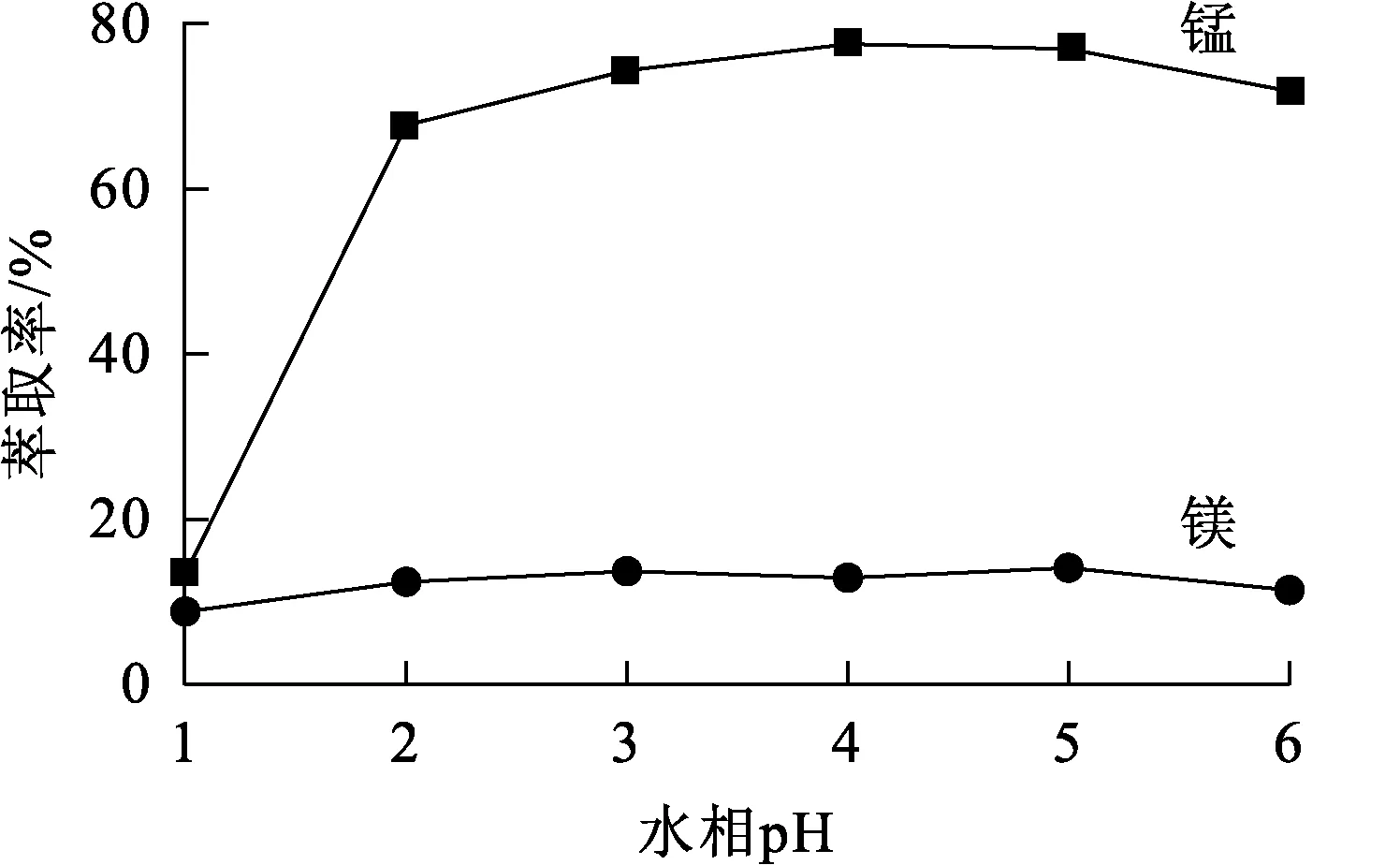
圖1 水相pH對錳、鎂萃取率的影響
2.1.2 萃取時間對錳、鎂萃取率的影響
相比Vo/Va=1.5/1,萃取劑體積分數30%,有機相皂化率30%,溫度25 ℃,水相pH=4.6,振蕩時間對萃取的影響試驗結果如圖2所示。可以看出:萃取反應在相對較短時間內達到平衡,3 min后錳、鎂萃取率基本達最大并趨于穩定。
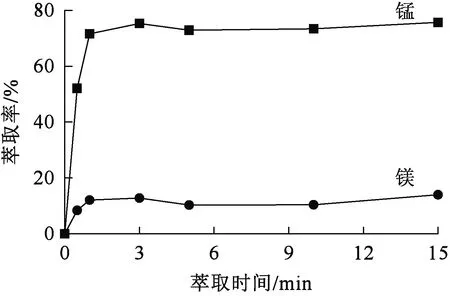
圖2 萃取時間對錳、鎂萃取率的影響
2.1.3 相比Vo/Va對錳、鎂萃取率的影響
萃取劑體積分數30%,皂化率30%,溫度25 ℃,水相pH=4.6,萃取時間15 min,相比Vo/Va對錳、鎂萃取率的影響試驗結果如圖3所示。
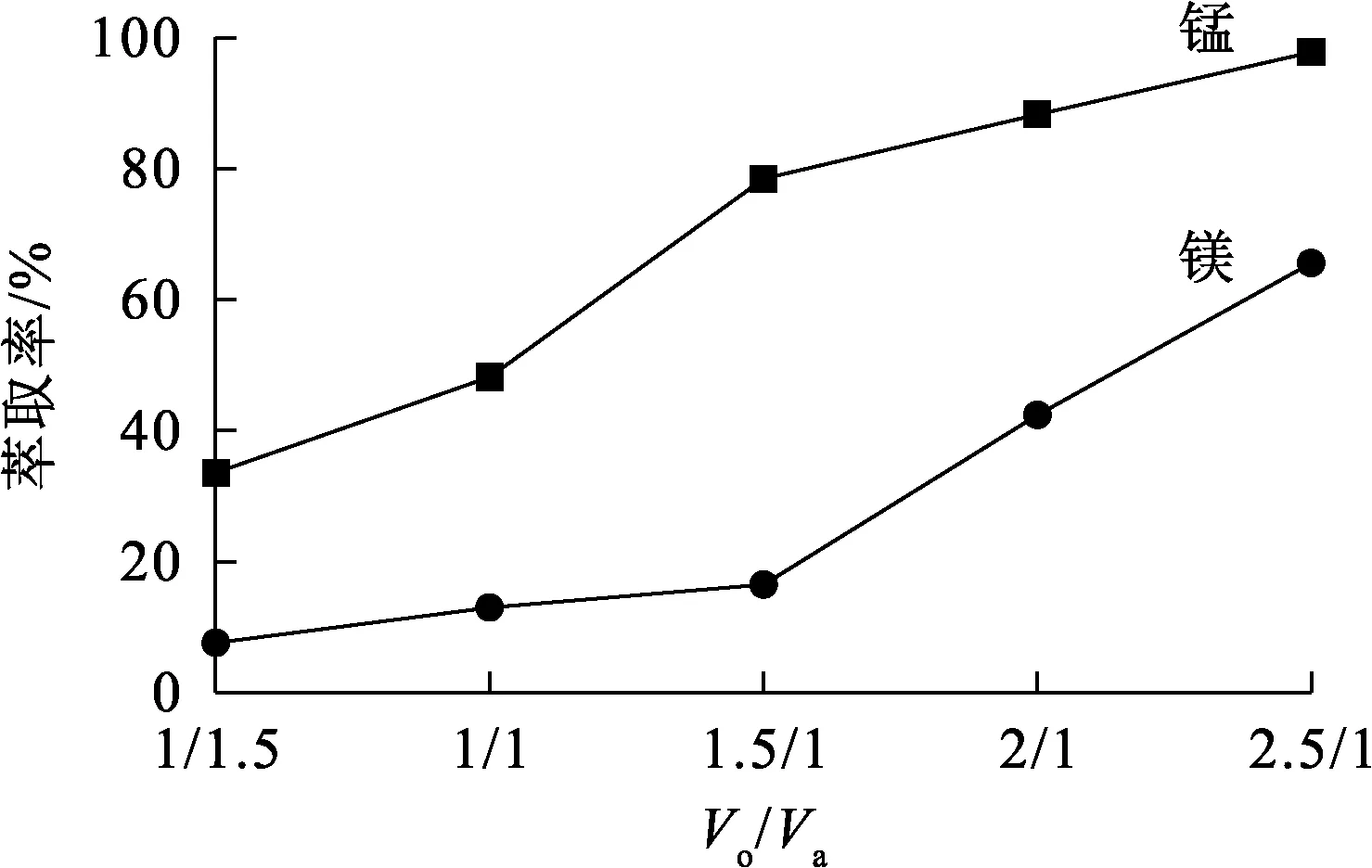
圖3 相比Vo/Va對錳、鎂萃取率的影響
由圖3看出:隨相比Vo/Va增大,錳、鎂離子萃取率均升高;Vo/Va在1.5/1以下時,隨Vo/Va增大,錳萃取率提升較快,鎂萃取率提升較慢;Vo/Va增至1.5/1以上后,水相中剩余錳質量濃度較低,萃取率提升較慢,過量的萃取劑繼續萃取鎂離子,導致鎂萃取率迅速提升。為了使錳、鎂離子更好地分離,確定適宜相比Vo/Va=1.5/1。
2.1.4 萃取劑體積分數對錳、鎂萃取率的影響
相比Vo/Va=1.5/1,有機相皂化率30%,溫度25 ℃,水相初始pH=4.6,萃取時間15 min,萃取劑體積分數對錳、鎂萃取率的影響試驗結果如圖4所示。
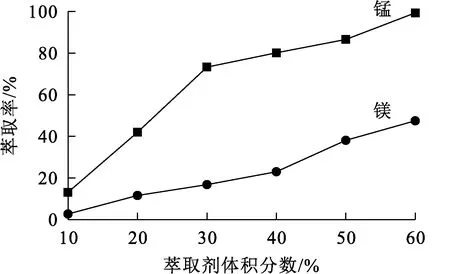
圖4 萃取劑體積分數對錳、鎂萃取率的影響
由圖4看出:隨萃取劑體積分數增大,錳、鎂萃取率都有較大幅度提高;萃取劑體積分數大于30%后,繼續增大萃取劑體積分數對錳萃取率影響不大,而鎂萃取率升高明顯。過大的萃取劑體積分數對錳、鎂分離沒有益處,且會造成萃取劑浪費和有機相黏度增大,以及體系乳化,綜合考慮,確定萃取劑體積分數以30%為宜。
2.1.5 有機相皂化率對錳、鎂萃取率的影響
相比Vo/Va=1.5/1,萃取劑體積分數30%,溫度25 ℃,水相pH=4.6,萃取時間15 min,有機相皂化率對錳、鎂萃取率的影響試驗結果如圖5所示。
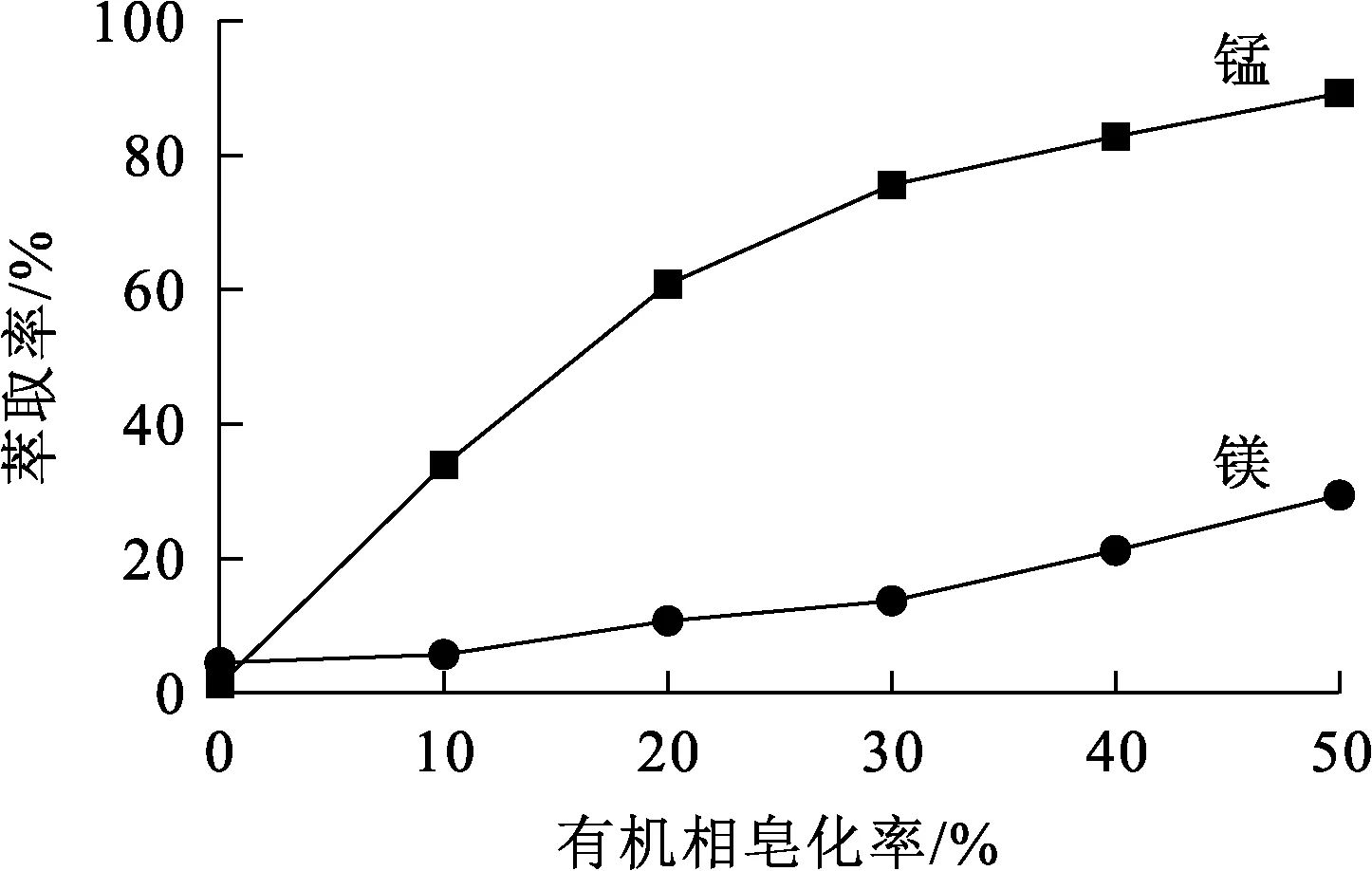
圖5 有機相皂化率對錳、鎂萃取率的影響
由圖5看出,隨有機相皂化率升高,錳、鎂萃取率均提高,錳萃取率提高幅度較大,鎂萃取率提高幅度較小:皂化率為0時,萃取劑對錳、鎂離子基本不萃取;皂化率為30%時,錳萃取率為75.6%,鎂萃取率為13.7%,錳、鎂分離效果較為理想;皂化率過大時,會導致體系乳化,造成分相困難。因此,確定有機相皂化率以30%為宜。
2.1.6 硫酸銨質量濃度對錳、鎂萃取率的影響
實際的電解錳陽極液中存在大量硫酸銨,質量濃度一般為80~100 g/L。試驗考察了硫酸銨質量濃度對錳、鎂萃取率的影響。試驗條件:相比Vo/Va=1.5/1,萃取劑體積分數30%,有機相皂化率30%,溫度25 ℃,水相pH=4.6,萃取時間15 min。試驗結果如圖6所示。可以看出,硫酸銨質量濃度對錳、鎂萃取率影響不大。
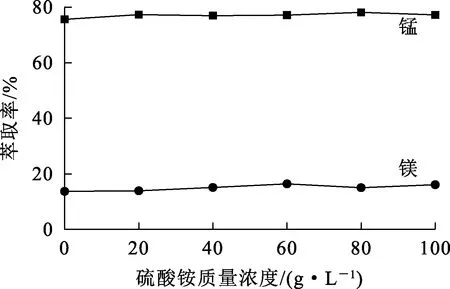
圖6 硫酸銨質量濃度對錳、鎂萃取率的影響
2.2 反萃取
2.2.1 相比的影響
用1 mol/L硫酸溶液作反萃取劑,有機相中錳離子質量濃度為8.89 g/L,溫度25 ℃,反萃取時間15 min,相比Vo/Va對反萃取的影響試驗結果如圖7所示。
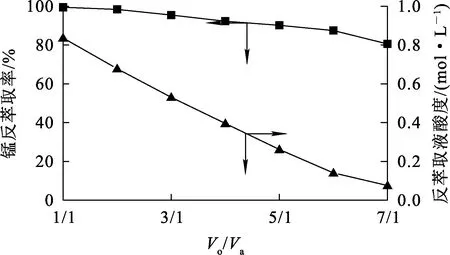
圖7 相比Vo/Va對錳反萃取率和反萃取液酸度的影響
由圖7看出:隨反萃取相比Vo/Va增大,錳反萃取率緩慢下降,水相剩余酸度大幅下降至0.076 mol/L。相比Vo/Va較低時,錳反萃取率較高,但反萃取后液剩余酸度高,中和剩余酸和蒸發結晶會造成資源和能源浪費;相比Vo/Va較高時,錳反萃取率略低,但反萃取液剩余酸度低,水相錳濃度高,對蒸發結晶有利。
2.2.2 反萃取酸度的影響
相比Vo/Va=1/1,溫度25 ℃,反萃取時間15 min,反萃取劑硫酸溶液濃度對反萃取影響試驗結果如圖8所示。
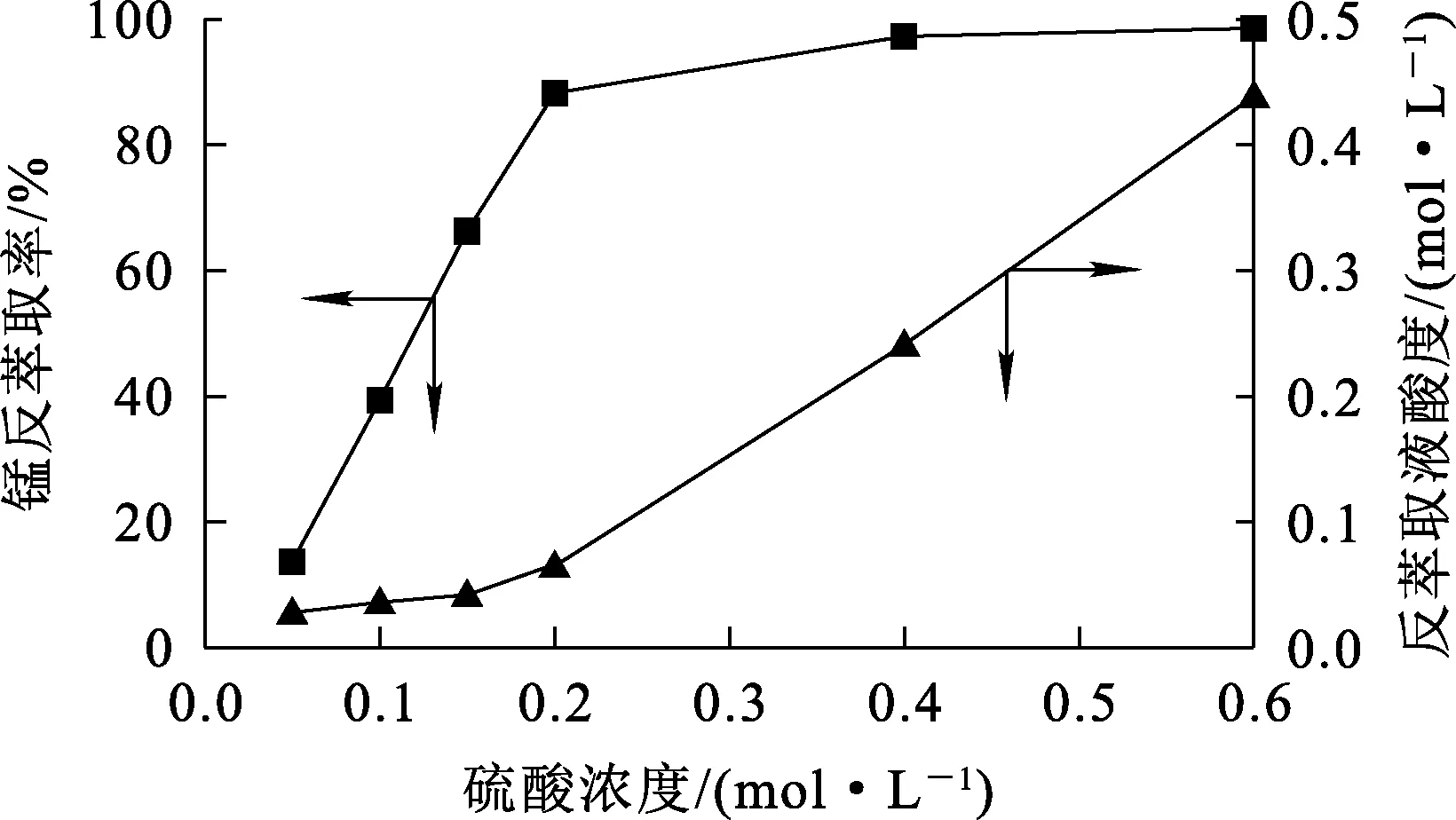
圖8 硫酸濃度對錳反萃取率和反萃取液酸度的影響
由圖8看出:錳反萃取率和水相剩余酸度隨硫酸濃度升高而升高;硫酸濃度增至0.2 mol/L時,錳反萃取率達88.3%,繼續增大硫酸濃度,錳反萃取率提高幅度不大,而反萃取液酸度會大幅提高。體積分數30%、皂化率30%的萃取劑可以負載9 g/L(0.164 mol/L)左右錳,試驗結果與理論預期符合。
2.3 高純硫酸錳的制備
在優化條件下進行萃取,被萃取液為模擬電解錳陽極液。錳萃取率77.9%,鎂萃取率13.7%,有機相中錳質量濃度為8.77 g/L,鎂質量濃度為1.93 g/L,用40 g/L(Mn2+)硫酸錳溶液進行一次洗滌[23],洗滌后有機相中錳質量濃度為10.67 g/L,鎂質量濃度為0.043 g/L。對負載有機相進行反萃取,反萃取劑硫酸濃度為1 mol/L,相比Vo/Va=6/1,此時錳反萃取率為82.4%,反萃取液中錳離子質量濃度為52.76 g/L,鎂離子質量濃度為0.21 g/L。用高純碳酸錳中和一次反萃取液pH至3.0以上,進行第二次萃取,錳萃取率為87.1%;對負載有機相進行反萃取,反萃取液中錳質量濃度為46.66 g/L,錳反萃取率為98.9%,反萃取劑硫酸濃度0.141 mol/L。
反萃取液用活性炭吸附除油,用高純碳酸錳中和pH至4.0~4.2,過濾掉多余的碳酸錳,緩慢蒸發濾液,濃縮結晶得到硫酸錳晶體,其中來自陽極液的錳和碳酸錳的錳比例為6/1。硫酸錳晶體在105 ℃下干燥8 h得到一水硫酸錳產品。經ICP-MASS測定,產品中錳質量分數為32.42%,鎂質量分數為9.32×10-6,質量符合《電池用硫酸錳》(HG/T 4823—2015)一等品要求。
3 結論
用一種新型萃取劑A從模擬電解錳陽極液中萃取錳,然后用硫酸溶液反萃取得到較純硫酸錳反萃取液,吸附去除殘留有機相后蒸發結晶,得到電池用硫酸錳。該方法簡單易行,錳、鎂萃取分離效果較好,錳回收率在70%以上,電解液中硫酸銨的存在對錳、鎂分離影響不大。所得一水硫酸錳產品,錳、鎂質量分數分別大于32%和低于1.5×10-5,質量符合《電池用硫酸錳》(HG/T 4823—2015)一級品要求。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
