四氧化三錳粒度及微觀形貌的控制
2022-02-18 08:03:08農(nóng)艷莉李春流楊茂峰萬(wàn)維華閆冠杰廖偉鋒
濕法冶金 2022年1期
農(nóng)艷莉,李春流,阮 籍,楊茂峰,萬(wàn)維華,閆冠杰,廖偉鋒
(南方錳業(yè)集團(tuán)有限責(zé)任公司崇左分公司,廣西 崇左 532200)
四氧化三錳(Mn3O4)為鋰離子電池正極材料錳酸鋰(LiMn2O4)的優(yōu)質(zhì)原料[1-2]。研究發(fā)現(xiàn),Mn3O4與LiMn2O4的晶體結(jié)構(gòu)相似,均為尖晶石結(jié)構(gòu)。以Mn3O4為錳源制備LiMn2O4,結(jié)構(gòu)變化較小,所得材料性能更優(yōu)異,所制備錳酸鋰電池的克比容量、循環(huán)壽命及高溫性能更優(yōu)[3-5]。
用Mn3O4制備LiMn2O4,Mn3O4的粒度、晶體形貌、振實(shí)密度及雜質(zhì)含量等指標(biāo)對(duì)LiMn2O4的性能具有重要影響,而Mn3O4的制備方法對(duì)其指標(biāo)影響較大。Mn3O4的制備方法主要有焙燒法、還原法、氧化法、電解法等。目前多采用金屬錳粉懸浮液氧化法[6-8],此法較為成熟,但生產(chǎn)成本高,雜質(zhì)含量高[9]。如何改進(jìn)制備工藝,降低雜質(zhì)含量,是目前的研究熱點(diǎn)。近年來(lái),用硫酸錳溶液在堿性介質(zhì)中不經(jīng)電解直接氧化制備Mn3O4得到廣泛研究,此法成本低,易操作,原料來(lái)源廣,是一種高效制備Mn3O4的新工藝[10-12]。
試驗(yàn)以一水硫酸錳為原料,采用一步氧化法制備類球形Mn3O4,以獲得微觀形貌、粒度分布更優(yōu)的Mn3O4產(chǎn)品。
1 試驗(yàn)部分
1.1 試劑與儀器設(shè)備
試劑:一水硫酸錳(純度>98%);氨水,氫氧化鈉,聚乙二醇1000(PEG),十二烷基三甲基氯化銨(DTAC),十二烷基苯磺酸鈉(SDBS),均為分析純。
儀器設(shè)備:YZCMR-10(M)型反應(yīng)釜,TYPE Y1991-2型空壓機(jī),SHZ-DⅢ型真空泵,DHG9076A型干燥箱,LA-300型激光粒度儀,NOVA 4000e型比表面分析儀,F(xiàn)ZS4-4B型振實(shí)密度測(cè)定儀,日立3400N掃描電子顯微鏡,D8 ADVANCE型X射線粉末衍射儀。
1.2 試驗(yàn)原理
根據(jù)圖1所示的φ-pH關(guān)系,錳鹽濕法制備Mn3O4的途徑主要有2種:1)Mn2+直接氧化生成Mn3O4,簡(jiǎn)稱一步氧化法;2)Mn2+首先生成Mn(OH)2,再經(jīng)氧化獲得Mn3O4,簡(jiǎn)稱二步氧化法。一步氧化法流程短,成本較低,但溶液體系pH、溫度、表面活性劑等對(duì)顆粒的形成及微觀結(jié)構(gòu)影響較大。一步氧化法制備Mn3O4顆粒需經(jīng)過(guò)形核、生長(zhǎng)、聚結(jié)、團(tuán)聚等過(guò)程[12]。根據(jù)晶體生長(zhǎng)的Lamer模型(圖2),在第Ⅰ階段,開(kāi)始反應(yīng)生成Mn3O4溶質(zhì)不斷積累,未形成沉淀;第Ⅱ階段為形核階段,隨著Mn3O4越來(lái)越多,達(dá)到形核所需的最低過(guò)飽和濃度時(shí),便開(kāi)始形成晶核,而形核消耗Mn3O4的速率超過(guò)供給速率,Mn3O4濃度不斷下降,進(jìn)入第Ⅲ階段即生長(zhǎng)階段[13-15]。濕法合成粉體材料的條件直接影響顆粒的生長(zhǎng)。根據(jù)Gibbs-Wulff晶體平衡形態(tài)理論,多面體的各個(gè)晶面的生長(zhǎng)速率各不相同,不同晶面可以通過(guò)選擇配離子、吸附簡(jiǎn)單離子或有機(jī)化合物分子(如表面活性劑)來(lái)改變其晶面的自由能,進(jìn)而促進(jìn)或抑制該晶面的生長(zhǎng)速度,最終可實(shí)現(xiàn)對(duì)顆粒尺寸、結(jié)構(gòu)形貌的控制。
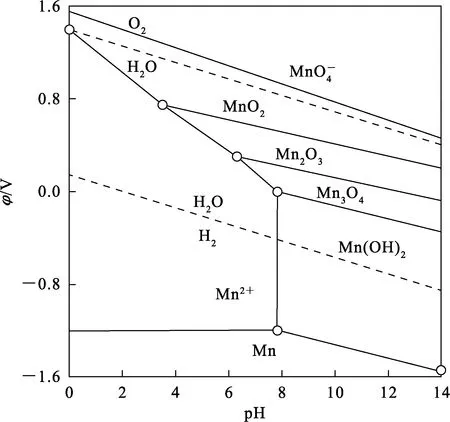
圖1 Mn-H2O系的φ-pH關(guān)系
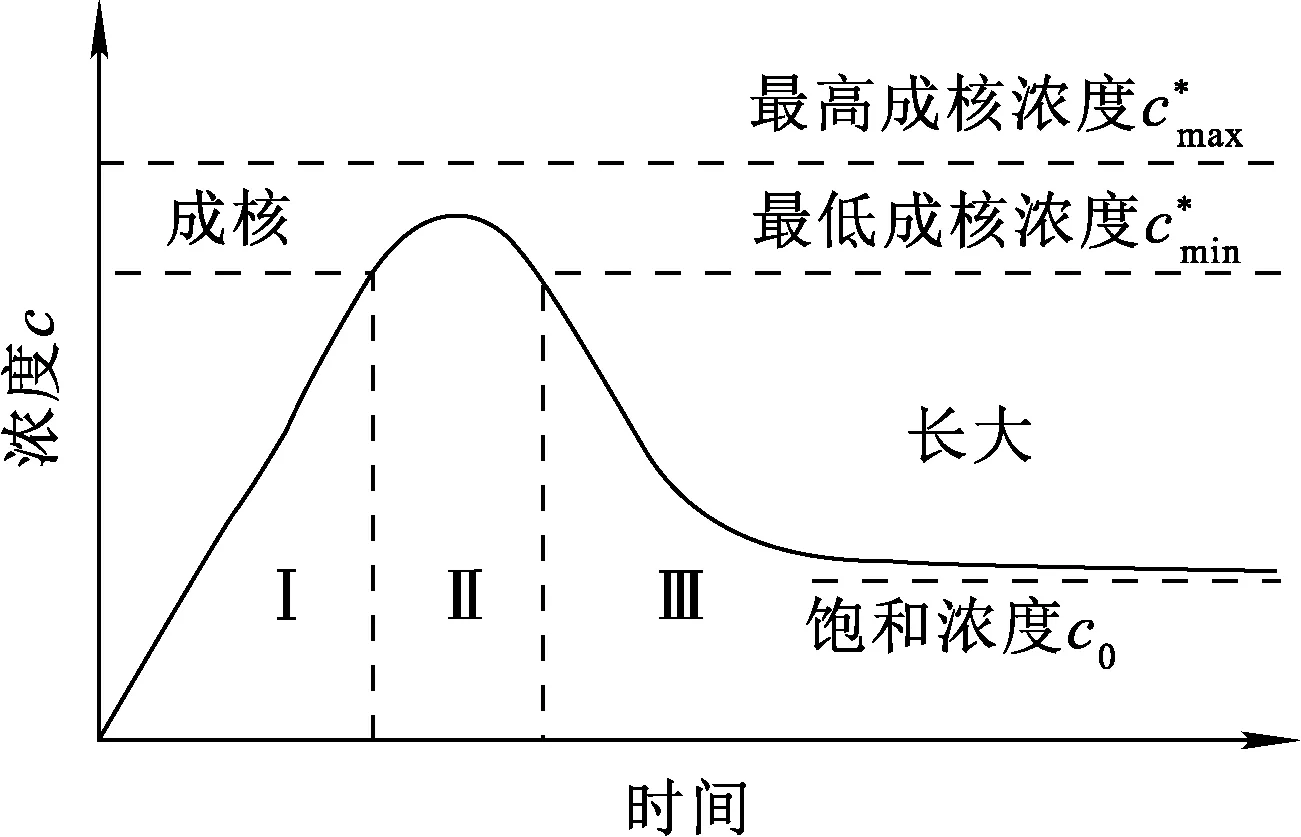
圖2 顆粒形成的Lamer模型
1.3 試驗(yàn)方法
配制2 000 mL濃度為2.0 mol/L的硫酸錳溶液并置于燒杯中,再向燒杯中加入一定量表面活性劑;將去離子水加入到反應(yīng)釜中作為底液,沒(méi)過(guò)攪拌槳;用蠕動(dòng)泵將硫酸錳溶液加入到反應(yīng)釜中,同時(shí)向反應(yīng)釜通入空氣進(jìn)行氧化。反應(yīng)過(guò)程中,攪拌速度為500~700 r/min,常壓,溫度45~75 ℃。反應(yīng)過(guò)程中加入堿以中和反應(yīng)產(chǎn)生的H+,將pH控制在6.4~7.64范圍內(nèi)。硫酸錳溶液加液完畢后,繼續(xù)加氨水控制pH,并穩(wěn)定攪拌12 h。最后過(guò)濾,用去離子水洗滌濾餅數(shù)次后放入烘箱中于120 ℃下干燥6 h,得到最終產(chǎn)品Mn3O4。試驗(yàn)所用表面活性劑有聚乙二醇1000(PEG)、十二烷基三甲基氯化銨(DTAC)和十二烷基苯磺酸鈉(SDBS)。
2 試驗(yàn)結(jié)果與討論
2.1 單因素試驗(yàn)
2.1.1 表面活性劑種類對(duì)Mn3O4顆粒形貌的影響
添加不同表面活性劑所得Mn3O4顆粒的SEM分析結(jié)果如圖3所示。

a—未加表面活性劑;b—PEG-1000;c—DTAC;d—SDBS。
由圖3(a)看出:未加入表面活性劑所得Mn3O4顆粒較小,形貌不規(guī)則,存在團(tuán)聚現(xiàn)象。一步氧化法制備Mn3O4,反應(yīng)生成顆粒過(guò)程中,晶核、微粒之間相互碰撞而團(tuán)聚生長(zhǎng)形成大顆粒,而攪拌剪切作用較弱時(shí),更有利于顆粒團(tuán)聚。晶體形核與長(zhǎng)大過(guò)程,不同晶面生長(zhǎng)速率各不相同,容易導(dǎo)致顆粒形貌不規(guī)則,無(wú)法形成球狀體,且極易受到表面活性劑等有機(jī)化合物分子的影響。表面活性劑對(duì)Mn3O4顆粒的微觀形貌及粒徑分布影響顯著(圖3(b)~(d)):以PEG-1000為表面活性劑,Mn3O4的一次顆粒團(tuán)聚形成較大的二次顆粒,結(jié)構(gòu)形貌不規(guī)則;以DTAC為表面活性劑,Mn3O4呈現(xiàn)不規(guī)則棒狀和類球形狀形貌,顆粒大小分布不均勻;以SDBS為表面活性劑,Mn3O4的微觀形貌呈較規(guī)則的類球形。
3種表面活性劑的分子結(jié)構(gòu)、溶解性和官能團(tuán)結(jié)合離子能力各不相同(圖4):PEG-1000屬于非離子型表面活性劑,具有較長(zhǎng)的疏水鏈,吸附于顆粒表面后,其親水基團(tuán)伸向液體介質(zhì),形成空間位阻效應(yīng)使顆粒相互排斥,但過(guò)量后易在粉體顆粒間架橋?qū)е滦跄龍F(tuán)聚;DTAC為陽(yáng)離子表面活性劑,在溶液中離解形成帶有疏水基的陽(yáng)離子C15H34N+,吸附在Mn3O4晶體表面形成疏水膜,控制晶體生長(zhǎng)速率,使不同晶面生長(zhǎng)速率不同,從而控制晶體形貌[16];SDBS為陰離子型表面活性劑,在水溶液中會(huì)組裝成球形膠束,非極性烴鏈為疏水基朝向里面,苯磺酸根為親水基朝向外面,Mn2+被吸附在SDBS膠束周圍并以此膠束為模板生長(zhǎng)[17-18]。
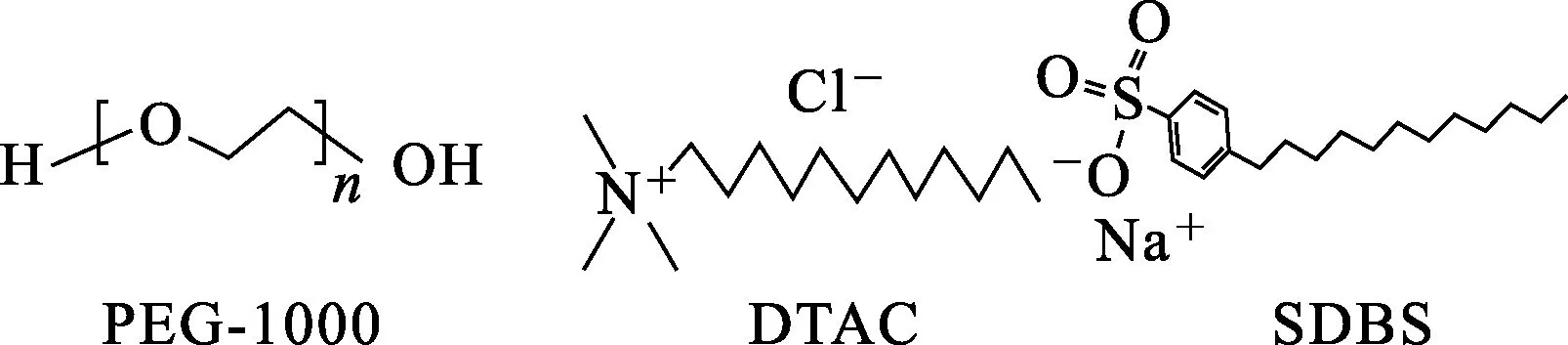
圖4 PEG-1000、DTAC和SDBS的結(jié)構(gòu)式
2.1.2 SDBS加入量對(duì)Mn3O4粒度、振實(shí)密度及Mn質(zhì)量分?jǐn)?shù)的影響
控制其他條件不變,以SDBS作表面活性劑,考察SDBS加入量對(duì)Mn3O4粒度、振實(shí)密度及Mn質(zhì)量分?jǐn)?shù)的影響,試驗(yàn)結(jié)果見(jiàn)表1。

表1 SDBS加入量對(duì)Mn3O4粒度、振實(shí)密度及Mn質(zhì)量分?jǐn)?shù)的影響
由表1看出:隨SDBS加入量增加,Mn3O4粒度增大,振實(shí)密度先增大后減小;SDBS加入量為1.0 g時(shí),Mn3O4粒度分布窄,振實(shí)密度達(dá)2.39 g/cm3,顆粒呈類球形(圖3(d));SDBS加入量繼續(xù)增加,粒度變化趨向平緩,振實(shí)密度有所減小。SDBS膠束作用明顯影響了Mn3O4的結(jié)晶和取向性生長(zhǎng),在恒定攪拌強(qiáng)度下,SDBS膠束的誘導(dǎo)更有利于Mn3O4晶體生長(zhǎng)形成類球形顆粒。在一定范圍內(nèi),SDBS加入量越大,類球形成型效果越明顯;但SDBS加入量過(guò)大,顆粒表面包裹層過(guò)厚,不利于顆粒的繼續(xù)生長(zhǎng)和微孔的填充,振實(shí)密度降低。
2.1.3 中和劑對(duì)Mn3O4顆粒形貌的影響
硫酸錳濃度2.0 mol/L,加料速度10 mL/min,反應(yīng)溫度60 ℃,攪拌速度600 r/min,SDBS加入量1.0 g,分別以氫氧化鈉和氨水為中和劑制備Mn3O4,中和劑對(duì)Mn3O4顆粒形貌的影響試驗(yàn)結(jié)果如圖5所示。

a—?dú)溲趸c;b—氨水。
由圖5看出,中和劑不同,Mn3O4顆粒形貌完全不同:以氫氧化鈉為中和劑,Mn3O4顆粒不規(guī)則,表面疏松,振實(shí)密度為1.27 g/cm3;以氨水為中和劑,Mn3O4顆粒呈規(guī)則的類球形顆粒,表面致密,振實(shí)密度為2.41 g/cm3。這主要是因?yàn)椋彼隗w系中起到中和酸和配合作用:一方面中和反應(yīng)過(guò)程中產(chǎn)生的H+;一方面與錳離子形成錳銨配離子,對(duì)Mn3O4的成核速率起到抑制作用,保證Mn3O4的成核、長(zhǎng)大,有利于得到形貌規(guī)則、分布均勻的類球形顆粒。
2.1.4 體系pH對(duì)Mn3O4粒度及形貌的影響
其他條件不變,用氨水控制體系pH,所得Mn3O4的SEM表征結(jié)果如圖6所示。

a—pH=6.5;b—pH=7.0;c—pH=7.5。

2.2 正交試驗(yàn)
粉體顆粒的微觀形貌、粒度分布等指標(biāo)與溶液濃度、攪拌強(qiáng)度、反應(yīng)溫度、配合強(qiáng)度等相關(guān)。以十二烷基苯磺酸鈉(SDBS)為表面活性劑,采用3水平4因素(34)設(shè)計(jì)正交試驗(yàn)方案,試驗(yàn)因素條件及結(jié)果見(jiàn)表2。可以看出:對(duì)Mn3O4粒度等指標(biāo)的影響順序?yàn)镈>C>B>A,即氨錳物質(zhì)的量比影響最大,其次是反應(yīng)溫度、攪拌速度,影響最小的是硫酸錳濃度;其中最優(yōu)條件為A1B1C3D3,即硫酸錳濃度1.0 mol/L,攪拌速度500 r/min,反應(yīng)溫度75 ℃,氨錳物質(zhì)的量比2.6/1。

表2 正交試驗(yàn)因素、條件及結(jié)果
2.3 最優(yōu)條件下的試驗(yàn)
在上述最優(yōu)條件下進(jìn)行試驗(yàn),對(duì)所得Mn3O4進(jìn)行XRD、SEM、粒度分布、雜質(zhì)含量等分析,結(jié)果見(jiàn)表3及圖7~9。可以看出:最優(yōu)條件下制備的Mn3O4為標(biāo)準(zhǔn)Mn3O4(PDF:24-0734),沒(méi)有其他錳氧化物雜質(zhì)存在,純度高,結(jié)晶度高;具有規(guī)則的類球形微觀形貌,結(jié)晶良好,致密性好,粒度符合正態(tài)分布,中位粒徑D50=11.24 μm,分布窄,顆粒大小均勻;Mn質(zhì)量分?jǐn)?shù)為70.58%,S質(zhì)量分?jǐn)?shù)為0.18%,其他雜質(zhì)質(zhì)量分?jǐn)?shù)都在5×10-5以下,振實(shí)密度為2.47 g/cm3,比表面積為0.87 m2/g。

表3 樣品的指標(biāo)分析

圖7 Mn3O4樣品的XRD圖譜

圖8 Mn3O4樣品的SEM照片
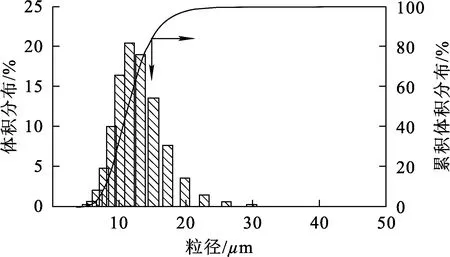
圖9 Mn3O4樣品的粒度分布
3 結(jié)論
以硫酸錳為原料,采用一步氧化法可制備四氧化三錳。表面活性劑對(duì)四氧化三錳微觀形貌影響顯著:以陰離子型表面活性劑SDBS為添加劑,所制備的Mn3O4微觀形貌和粒度分布更好;中和劑氨水的加入及控制體系pH為7.5,可獲得球形度及粒度分布更好的Mn3O4。適宜條件下,所制備Mn3O4產(chǎn)品粒度為11.24 μm,顆粒呈明顯類球形,振實(shí)密度高,為2.47 g/cm3,比表面積小,雜質(zhì)含量低,可用作高性能錳酸鋰制備的前驅(qū)體材料。
