離子嵌入電化學反應機理的理解及性能預測:從晶體場理論到配位場理論
2022-02-19 08:39:24王佳民蒲博偉李銘清寧芳華李亞捷施思齊
儲能科學與技術 2022年2期
王 達 ,周 航 ,焦 遙 ,王佳民 ,施 維 ,蒲博偉 ,李銘清 ,寧芳華 ,任 元 ,喻 嘉 ,李亞捷 ,李 彪 ,施思齊 ,,5
(1上海大學材料科學與工程學院,上海 200444;2上海大學材料基因組工程研究院,上海 200444;3上海大學可持續能源研究院,上海 200444;4法蘭西學院,法國 巴黎UMR 8260;5之江實驗室,浙江 杭州 311100)
離子嵌入電化學是推動電化學儲能技術進入現代化的關鍵因素之一[1-3]。在典型離子嵌入電化學裝置中,電極較為充裕的通道空間既能夠儲存大量離子又有利于單價/多價離子快速嵌入/脫出。更重要的是,遷移離子的脫嵌僅導致主體結構產生微小的結構變化,以此確保了電極優異的倍率性能和循環穩定性[4-5]。因此,以鋰離子電池為代表的離子嵌入電化學裝置在便攜式電子產品中占據主導地位,同時離子嵌入電化學對新型儲能系統發展有著突出的貢獻,是清潔能源電網和電氣化交通的主要儲能候選者[6-10]。自20世紀70年代以來,鋰離子電池和其他單價/多價金屬離子電池(例如Na+、K+、Ca2+、Mg2+、Zn2+、Al3+)的電化學性能改進已成為研究熱點[11-13]。
單價/多價金屬離子電池的充放電循環反應主要包括電極上進行的電化學嵌入/脫出反應傳導電荷,電解質在兩個電極之間來回穿梭傳導離子兩個關鍵過程[14-15]。遷移離子的脫嵌與電子的傳輸共同導致電極發生氧化還原反應,最終引起氧化還原中心的電子結構發生變化[5]。其中,離子嵌入電化學中大多數正極是過渡金屬化合物(TMXs,TM=過渡金屬,X=O、S、N、C等),單價/多價金屬離子的嵌入/脫出伴隨著TM的氧化還原。本質上講,晶體基本結構單元(晶體原子和配位環境)的電子結構決定了晶體的物理和化學性質。上述正極材料的電子結構則取決于TM在不同配位環境下的d軌道分布/占據特性[16],并受到配位場的嚴格控制。此外,在離子嵌入過程中電子的注入也會進一步影響材料的配位環境[17-18]。因此,基于配位場理論建立材料局域環境對離子嵌入過程中的電壓平臺、相結構穩定性和比容量等性能的影響機理至關重要[19-21]。例如,Goodenough 教授[22]總結了正負極體系費米能級位置對電池嵌入電壓的決定關系,一般可將電池的開路電壓(Voc)用正極(μC)和負極(μA)之間的化學電位差(Δμ)表示:Voc=(μA-μC)/zF,其中z是電極之間轉移的電荷,F是法拉第常數,μC或μA的能量值可能對應于一個流動電子帶中的費米能級[23],而這些費米能級的變化起源于分子軌道分裂程度的變化。因此,可以首先通過晶體場論判斷中心原子分裂,再根據對稱性匹配原則、最大重疊原則和能量相近三大原理推導出中心原子軌道與配體形成的雜化分子軌道,進而判斷費米能級處電子的分布狀態與能級位置。因此,配位場理論的引入將為理解離子嵌入過程與電化學性能之間的相互作用機制提供了系統而獨特的視角,有助于進一步提出設計新型單價/多價金屬離子電池的理論策略。
由此,我們先將配位場理論的發展進行了全面的梳理。配位場理論是在晶體場理論的基礎上發展而來的。晶體場理論的基本思想由Becquerel 提出:絡合物中的金屬離子受到配體電場的影響。同年,Bethe[24]將該想法明確地表述為具體的理論,并且通過對稱概念研究了晶體場的對稱性和強度如何影響氣態金屬離子的電子能級。1935 年,Van Vleck[25]證明晶體場理論與分子軌道理論具有一致性,并將其成功應用于化學領域。然而,晶體場理論是在點電荷模型的基礎上推導出來的,它忽略了中心原子和配體間的共價成鍵,因此很難對化合物的性質給出定量的解釋。對此,分子軌道理論能夠給出更為合理的解釋,但要做精確的、非經驗的理論處理卻碰到了數學上的困難。在這種情況下,解決辦法是在晶體場理論的基礎上,將共價成鍵作用包含進去,同時引進晶體場參數。這種經過修飾的晶體場理論稱為配位場理論,顯然它是晶體場理論和分子軌道理論的結合[26-27]。自1952年起,配位場理論經過多年的發展已成為研究含TM 配合物系統中微觀結構、熱力學性質、磁性等問題的基礎,并廣泛應用于熱力學、礦物學以及電化學等領域[28]。盡管配位場理論的提出比離子嵌入電化學的發現早了近20年,但是隨著對離子嵌入電化學中的電荷存儲/轉移機制理解的逐步深入[29-30],如我們于2016年首次梳理了多尺度計算在理解電池材料上述機制中的作用[31],再加上第一性原理(FP/DFT)計算方法[32-33]和軟件[34]進步的推動,使得配位場理論在材料電化學性質研究和預測的應用中具有實用性和可擴展性,也讓配位場理論成為離子嵌入式電化學研究領域中不可缺少的一部分[35-36],如圖1 中總結所示。本論文中,我們結合第一性原理計算和配位場理論分析,將電極材料的局部化學配位、分子軌道占據特性與其電化學性質聯系起來,推導出了調控電化學性能的模型/理論,如離子嵌入電壓平臺、相結構穩定性等,這將為新型高性能電極材料的設計提供理論指導。本論文不僅能滿足初學者對配位場理論基礎知識的需求,還涵蓋了具有豐富工作經驗的化學工作者關注的配位場理論在離子嵌入電化學領域中的發展及改進方向等需進一步思考的深化主題。
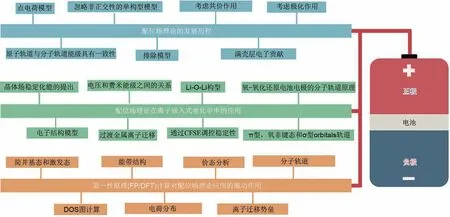
圖1 配位場理論的發展歷程(上),配位場理論在離子嵌入式電化學中的應用(中)以及第一性原理(FP/DFT)計算對配位場理論應用的推動作用(下)。上述因素共同推動了電池材料的性質研究和預測的發展Fig.1 Development history of ligand field theory(top),application of ligand field theory in ions-intercalation electrochemistry(middle),and driving factors of FP/DFT calculation for application of ligand field theory(bottom).Above factors have jointly promoted development of research and prediction on properties of battery materials
1 配位場理論的理論基礎
配位場理論于1952 年提出,其融合了晶體場的靜電作用和分子軌道的共價作用,成為研究熱力學、地質礦物學和電化學系統中的結構畸變、熱力學性質和磁性等諸多物理/化學問題的基礎。在單價/多價金屬離子嵌入電化學反應電池體系當中,正極材料通常為過渡金屬化合物(TMXs,TM=過渡金屬,X=O、S、N、C 等),離子的脫嵌往往伴隨著TM的氧化還原(也可能發生陰離子同時參與氧化還原反應現象,如本文2.3節討論)。晶體場理論則描述了局域配位環境對這些電極材料中TM-d 軌道能級的影響,并決定著電極費米能級、電荷轉移、離子輸運等性質的變化。配位環境對中心離子的作用起源于配體的靜電場,配體包括中心離子附近的陰離子或偶極基團,通常被視為點負電荷。就此,首先需對晶體場靜電作用的概念進行闡述。
晶體場效應取決于配體所產生靜電場的對稱性和強度,中心TM 離子受到的影響取決于周圍配體的類型、位置和對稱性。原子核周圍每個電子的位置和能量都可由波函數描述,即薛定諤波動方程的解。波函數描述了原子核周圍電子密度的空間分布,這與某一時刻在特定區域遇到電子的概率有關。電子的波函數可以寫成4 個獨立函數的乘積,其中的3個取決于電子的極坐標。這3個函數包括:與電子到原子核的徑向距離r相關的徑向函數,以及分別與角度θ和φ相關的兩個角函數。第4 個函數為自旋函數,與空間坐標r、θ和φ都無關。電子的總波函數可以寫為乘積

通過繪制表面來封閉角波函數的振幅。這些邊界表面就是原子軌道,每個軌道的波瓣都有正負符號,作為薛定諤波動方程的數學解。
1.1 基于晶體場理論的原子軌道分裂
盡管晶體場對中心離子d軌道相對分裂的定性影響很容易理解,但晶體場效應的定量計算是一個十分復雜的數學問題。首先需建立中心金屬離子受配體場作用的總晶體場勢的表達式,然后將這種晶體場勢對d 軌道的影響看作是一種微擾作用[37]。通過哈密頓量來描述擾動和相應的矩陣元素,就可以通過標準微擾理論找到晶體場中的波函數及其能級。此時中心原子的d軌道受到來自周圍分子(稱為配體)的電場的影響,導致軌道的簡并性減少。如果忽略電子自旋軌道耦合對總能量的貢獻,則哈密頓量為

根據微擾理論,式(2)中前兩項表示單電子在離子的位場中運動的哈密頓,用H0表示,這時已知自由離子中5個d軌道的能級是簡并的。式(2)中的后一項V'即為微擾,用H'表示[38]。d 軌道能級在晶體場中的分裂是由晶體場位能V'的微擾所引起。其中對于P個配位體微擾勢的表達式為

其中ρk(r)是rk/Rjk+1的縮寫,其中項Zkα(θj,φj)是評估角度(θj,φj)的一組數值,它描述了第j個配體的位置,項Zkα(θ,φ)表示金屬離子電子的角位置。由于晶體場勢V'(r,θ,φ)的應用,相對能量的變化程度Ek可以通過微擾理論來進行計算,通過久期方程求解,可以得到下列矩陣元


下面針對單價/多價金屬離子電池中最為常見的ML6(M=過渡金屬,L=配體)分子構型的電極材料d 軌道劈裂系數進行推導。ML6分子構型的電極材料主要為八面體構型,如LiCoO2、LiFeSO4等電極。八面體的d軌道的對角矩陣元為

正八面體屬于Oh群,d軌道分屬于兩個不同的不可約表示:eg(dz2,dx2-y2)和t2g(dxy,dxz,dyz)。將H'ii值與表S2對照可得

其中,〈r2〉和〈r4〉的值可以使用類氫Slater 軌道或Hartree-Fock 自洽場自由離子d 波函數來計算[39-41]。通常用符號ρ表示比率ρ2(r)/ρ4(r),并使用定義Dq=〈ρ4(r)〉/6=ZLe2〈r4〉/6R5。對于目前研究的復合體系,徑向積分參數ρ的經驗值一般定義為2.0[42]。所以式(6)=+6×Dq,式(7)=-4×Dq。
關于其他構型的推導詳見支撐材料S1 部分,可以得到不同晶體場下的d軌道能級分裂情況,可為后續配位場理論調控電化學性能提供理論基礎。
1.2 基于分子軌道理論構造分子軌道能級圖

下面以八面體ML6型分子為例來闡述基于分子軌道理論如何構造分子軌道能級圖。首先找出所有σ 軌道形成基的可約表示。中心原子M 與配體L 可形成的分子軌道(以及與L配體的原子軌道線性組合而成的函數)一定屬于6根M-L鍵所屬點群為基組進行對稱操作得到的可約表示。所以我們將分子點群的所有對稱操作作用到這組σ軌道上。應用恒等操作可以得到一組特征標,見表1。

表1 八面體構型分子形成σ鍵的可約化特征標Table 1 Reducible characteristics of Octahedral configuration molecules forming σ bonds
參考八面體點群Oh的特征標表(表S7),利用可約化公式化簡得到:Γ八面體=A1g+T1u+Eg。從上列分析可以看出,分屬于中心原子M 的原子軌道為:A1g(s);T1u(px, py, pz);Eg(dx2-y2,dz2)。故中心原子M 采取的雜化形式是d2sp3(d2指dx2-y2,dz2),這也是唯一可獲得的雜化方式。需要注意的是,MLn型分子除了會形成σ鍵之外,在單價/多價金屬離子電池電極材料中通常還會觀察到π鍵的存在,并且其可能對于離子嵌入電化學過程存在關鍵作用。例如,在目前較為熱門的富鋰金屬氧化物電極(TMOs)中,π成鍵分子軌道對氧離子的氧化還原活性產生了主導性影響。π 成鍵分子軌道處理的基本原理和σ 成鍵是一樣的,對ML6型結構π鍵的詳細推導見支撐材料S2部分(http://esst.cip.com.cn/CN/10.19799/j.cnki.2095-4239.2021.0652)。
綜上所述,對于八面體構型的ML6分子,配體群軌道與中心原子軌道按照對稱性匹配,最大重疊原則及能量相近原則形成σ鍵和π鍵,對應的分子軌道能級圖如圖2 所示。中心過渡金屬原子M 的5 個d 軌道分裂為eg(dx2-y2,dz2)和t2g(dxy, dxz, dyz)。其中eg將沿M-L鍵方向與配體p軌道直接重疊,形成能量較高的反鍵態e*g和能量較低的成鍵態eg帶,t2g(dxy, dxz, dyz)軌道與O-2p 雜化形成t*2g和t2g。由于配體群軌道能級低于金屬原子軌道能級,因此中心原子M 的d 電子填充在e*g和t*2g軌道上,標記為M(d)狀態。金屬原子的(n+1)s 和(n+1)p 軌道將與配體p(σ)態重疊,形成a1g和t1u帶。由于配體的電負性比中心原子更高,軌道能級更低,因此配體p(σ)態的電子主要位于eg、a1g、t1u和t2g的成鍵態上。由于它們的能量相近,所以通常將它們相互混合并標記為L(p)狀態。需要強調的是,對于分子軌道具體的雜化方式,除了上述具有單一的雜化方式外,還存在特定情況(詳細推導見支撐材料S2部分)需從能量的角度考慮其具體的雜化方式。
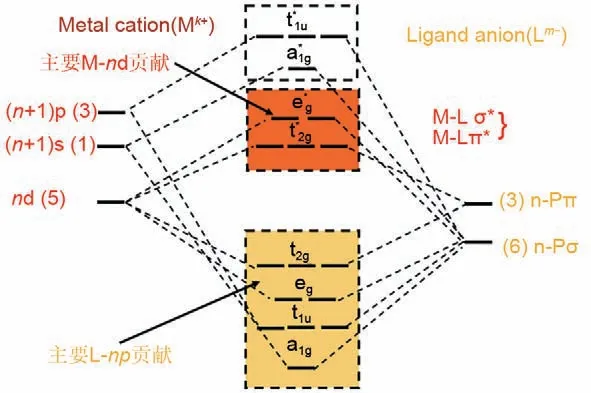
圖2 典型中心原子M與配體L軌道之間的σ鍵和π鍵的能帶結構圖。在單價/多價金屬離子電池的大多數離子嵌入式化合物中,過渡金屬陽離子Mk+與配體Lm-形成八面體配位,M的軌道與L的部分p軌道雜化形成σ鍵和π鍵,其中黃色部分代表由L的p軌道貢獻,紅色部分表示由M的d軌道貢獻Fig.2 Band structure diagram of σ bonding and π bonding between typical central atom M and ligand L orbital.In most ions-intercalation compounds in mono-/poly-valent metal ion batteries,transition metal cation Mk+forms an octahedral coordination with ligand Lm-.M orbitals hybridize with part of p orbitals of L to form σ bonds and π bonds.Yellow part represents contribution from p orbital of L,and red part represents contribution from d orbital of M
通過將上述基于分子軌道理論的配位場理論與第一性原理計算相結合,可獲得精確的分子軌道能級圖像,以此幫助我們把電極電化學性能研究從傳統基于材料體/表面的宏觀層面推向局域化學鍵的微觀層面,即從局部化學配位和雜化分子軌道來探討材料的物理/化學性質。由此可見,上述配位場理論分析為理解單價/多價金屬離子電池電極材料中局域電子結構特性提供一個新的視角。鑒于此,我們自主開發了“識別局部配位場構建晶體分子軌道能級程序(Construction of crystal molecular orbital energy level diagram by identifying local ligand fields, MOELD-LLF) V1.0”,實現了根據電極體系局域配位環境推導其原子軌道劈裂特性,并通過對稱性匹配原理確定體系雜化分子軌道能級排布這一流程的自動化。該程序目前已開源(訪問地址:https://gitlab.com/shuhebing/MOELD-LLF)。
2 配位場理論在離子嵌入電化學領域中的應用
基于上節對配位場理論基礎的全面梳理,本節將對離子嵌入電化學過程中決定電壓的費米能級計算模型、衡量相結構穩定性的晶體場穩定化能計算公式、調控陰離子氧化還原活性的理論模型等進行嚴格的推導。在此基礎上提出了剛性帶電極體系電壓平臺提升和不同周期材料晶體場穩定化能預測等一系列電極能量密度與循環穩定性能的改進策略,并成功設計出無過渡金屬B-C-F正極及嵌入式無鋰MX2正極兩種新型電極材料。
2.1 電極離子嵌入電壓平臺
在離子嵌入電化學反應中離子嵌入/脫出的電壓平臺和費米能級之間存在明確的關聯。對此,Goodenough 教授[22]也做出了系統的解釋,即將二次電池的開路電壓Voc定義為eVoc=μA-μC,其中e是單位電荷量,兩個電極為分別具有μA和μC電化學電位的電子導體(它們的費米能量EF),而μA或μC的能量分別對應了負極中鋰所處化學勢(和材料中Li-2s電子占據費米能級位置相關)或正極中TM離子氧化還原對的能量,如圖3(a)所示。其中電解液的最低未占據分子軌道(LUMO)和最高占據分子軌道(HOMO)的能量差是電解質的“窗口”。穩定的電池充放電循環性需滿足μC和μA在上述電解液電化學窗口內,否則可能會發生電解液分解等嚴重的副反應導致電池性能的衰減。對于負極材料,目前石墨與電解質溶劑反應在石墨表面上形成穩定的固體電解質界面層(SEI)為其操作提供了長壽命的穩定性。然而,通過SEI的緩慢鋰擴散可能導致石墨表面形成鋰枝晶和內部短路,導致災難性的安全隱患。Li4Ti5O12等負極材料電位雖位于LUMO 下方,不會形成SEI膜,提供長循環壽命,然而其工作電壓為1.5 V (vsLi/Li+),嚴重限制了其與正極匹配形成的開路電壓的拓寬。對于正極材料,商業化的材料主要有LiMO2(M=Co, Ni, Mn)、LiFePO4等,但是其工作電壓及其在電化學過程中的活性中心變價電子數等性能仍有待進一步的提升[43]。對此,Cherkashinin 等[23]將提升電壓的決定性因素分離為電子化學勢(Δμe)和離子化學勢(ΔμLi+)兩方面:Voc=ΔG/zF=(μA-μC)/zF=(Δμe+ΔμLi+)/zF,其中z是電極之間轉移的電荷,F是法拉第常數。由此可見,即使對于相同的電極材料,不同堿金屬或高價離子電化學勢、電負性、電離能等物理化學性質的不同會導致其脫嵌電壓平臺的不同。如Liu 等[44]發現相比同主族其他堿金屬/堿土金屬離子,Na+和Mg2+無法在石墨負極中脫嵌是由于其電離能及離子-基底耦合之間的競爭所導致。因此,調控電極材料本身離子嵌入能級(Δμe,即電極費米能級位置)對于改善上述離子無法嵌入問題起著極為重要的作用[圖3(b)]。考慮到電極材料費米能級(EF)的位置本質上是由雜化分子軌道的能級決定,深入探討EF附近的雜化分子軌道能級狀態對進一步提出電壓提升策略尤其關鍵。

圖3 (a)電解質的電化學窗口示意圖。最低未占據分子軌道(LUMO)和最高占據分子軌道(HOMO)的能量差為電解質窗口。ΦA和ΦC分別為負極和正極功函數,其中電壓Voc=ΔG/zF=(μA-μC)/zF,μA和μC分別為負極和正極處鋰的化學勢;(b)由正極、導電電解質和負極組成的典型單價/多價金屬離子(或堿/高價金屬離子)電池的電子能級示意圖Fig.3 (a)Schematic energy diagram of an aqueous electrolyte.Energy separation of lowest-unoccupiedmolecular-orbital(LUMO)and highest-occupied-molecular-orbital(HOMO)is electrolyte window.ΦA and ΦC indicate work functions of anode and cathode.Where voltage is Voc=ΔG/zF=(μA-μC)/zF,μA and μC represent chemical potentials of lithium at anode and cathode materials,respectively;(b)Schematic diagram of electronic energy levels of a typical mono-/poly-valent metal-ion battery composed of a positive electrode,a conductive electrolyte and a negative electrode
電極的工作電壓主要是由參與氧化還原反應的局部電子結構和電子狀態決定[22]。當電極材料在充放電過程中發生離子的脫嵌行為時,嵌入離子所帶來的電子將導致體系EF處簡并軌道能級發生進一步分裂。例如,常見單價/多價金屬離子電池中典型的LiFePO4、LiCoO2等八面體場電極材料EF處的雜化分子軌道通常為簡并的t*2g軌道,其在離子嵌入過程中將發生能級分裂,從而使得這些電極在離子嵌入前后的電子結構發生顯著的變化,如圖4(a)所示[45-46]。對于不同的單價/多價金屬離子電池,其不同價態離子嵌入導致填充電子數的不同不僅會影響過渡金屬中心原子周圍環境的電荷分布和晶格體積的變化,也將影響EF處簡并軌道能級的分裂程度,最終導致不同離子嵌入電極材料電化學性能的顯著差異。例如LiVPO4F材料在摻雜Na+、Mg2+、Al3+后提高材料的電子電導率的程度不同,其主要原因在于Na+、Mg2+、Al3+摻雜后對氧周圍的電荷分布影響不同,并且影響V-O八面體的體積膨脹程度,進而導致費米能級處的軌道能級位置以及電極電子電導率等性能發生明顯的變化[47]。

圖4 (a)LiFePO4正極材料的XPS價譜以及態密度(DOS)計算。對于Fe 3d,上下自旋部分DOS已被區分。導帶中的DOS繪制為虛線[46];(b)八面體MO6的分子軌道能級圖[48]Fig.4 (a)XPS valence spectrum and density of states(DOS)of LiFePO4 cathode.For Fe 3d,spin up and spin down partial DOS have been distinguished.DOS of conduction bands have been drawn as dotted lines;(b)Molecular orbital energy diagram for octahedral MO6
總體而言,調控用于單價/多價金屬離子電池的過渡金屬化合物電極材料電化學性能需同時考慮過渡金屬中心離子和配體電子性質的影響,這是由于過渡金屬化合物費米能級附近的分子軌道通常都為過渡金屬d 軌道和配體p 軌道之間的軌道重疊組成,導致局域在費米能級附近的成鍵態和反鍵態分別具有強配體p 軌道和金屬d 軌道的特性。由此可見,上述電極材料電化學性能將由過渡金屬、陰離子的種類以及晶體的結構共同決定。例如,對于最常見由八面體局域結構組成的電極材料,中心離子的d電子[標記為M(d)態]將劈裂為t*2g和e*2g軌道中,而配體p 軌道的電子(標記為L(p)狀態)主要局域在eg、a1g、t1u和t2g的成鍵態上,如圖4(b)所示[48]。由此,調控該類過渡金屬化合物電極電壓的方式本質上可劃分下列兩種情況:①調控中心離子,即通過替換不同電負性的中心離子直接改變體系EF附近金屬-配體鍵(M-L)的離子/共價特性來調控電壓;②配體調控,即利用聚陰離子基團存在的誘導效應間接改變M-L共價鍵強度進而調控電壓。下面來著重探討這兩種方式對電壓的影響。
2.1.1 調控中心離子
首先來探討不同電負性的過渡金屬中心離子對電極電壓的調控作用。電負性描述了原子或官能團向自身吸引電子(或電子密度)的趨勢,它是影響電極電化學電位的重要因素。從圖5(a)中可以看出,對于給定的聚陰離子,M3+/4+氧化還原對的放電電位值呈現出Ni>Co>Fe>Mn 的總體趨勢[49-52]。這是因為由于Mn和Fe的原子序數分別為25和26,Fe的電負性高于Mn,導致LiFeSiO4電極的電壓高于LiMnSiO4電極。然而,還存在對于M2+/3+氧化還原對的放電電位值與電負性趨勢并不完全一致的情況,例如,Fe 比Mn 具有更強烈地吸引電子能力,因此可能導致在電子嵌入或脫出過程中存在更高的能量消耗或釋放,然而實際卻發現與LiFePO4相比,LiMnPO4顯示出更高的放電電位。這是由Fe2+(3d6)和Mn2+(3d5)價電子數不同導致,即Fe2+(3d6)需要電子配對能量才能在t2g軌道中插入第六個電子,而Mn2+(3d5)可將電子填入更低能級的eg上自旋軌道[53],如圖5(b)所示[54]。由此可見,LiFePO4和LiMnPO4電壓的差異本質上來源于Mn2+中可用eg電子的能級狀態低于Fe2+中t2g電子的能級,最終導致LiMnPO4[55](4.13 V)比LiFePO4[56](3.43 V)具有更高的電位。綜上所述,過渡金屬中心離子電負性僅是關鍵影響因素之一,但并不能僅通過替換/摻雜中心離子來調控電壓,還需綜合考慮到其價電子數等其他性質的差異。
2.1.2 調控配體
接下來探討調控配體對過渡金屬化合物電極電壓的影響。對于特定的過渡金屬離子,不同的聚陰離子基團會導致嵌鋰電化學電位不同[57,58]。例如對于相同結構的Fe2(MoO4)3(3.0 V)和Fe2(SO4)3(3.6 V)電極,其雖有著相同的Fe2+/3+氧化還原中心對,但需通過改變聚陰離子基團中的Mo6+和S6+性質改變Fe2+/3+對的氧化還原能級,從而影響Fe-O 鍵的離子/共價特性。在Fe2(XO4)3(X=Mo 和S)結構中,FeO6八面體與XO4四面體共享角,提供了一個擴展的三維-O-Fe-O-X-O-Fe-O-連接通道。因此,與氧化物Fe2O3相比,更多的共價Mo-O 鍵通過誘導效應削弱了Fe-O 鍵的共價性,導致Fe2+/3+氧化還原能的降低和電壓的提高。而Fe2(SO4)3中更多的共價S-O鍵則進一步削弱了Fe-O共價特性,導致Fe2+/3+氧化還原能進一步地降低并進一步提升該電極電壓,如圖5(c)所示。從圖5(d)的軌道能級圖中可以看出,與同構Fe2(MoO4)3相比,Fe2(SO4)3中Fe2+/3+氧化還原能降低更多是由于共價S-O鍵相較于Mo-O削弱Fe-O共價的程度更大,共價鍵的減少削弱成鍵軌道和反鍵軌道之間的排斥力,使得反鍵軌道的能級更低,導致其相對電壓提高0.6 V[59]。因此,通過聚陰離子基團產生的誘導效應可顯著影響M-O 共價鍵的強度,進而影響電極電壓[60-61]。上述調控策略雖可以有效提高電極電壓并發現了電壓與費米能級存在的密切關系,但兩者之間的關系無法定量化,根本原因在于這類過渡金屬化合物正極材料通常具有八面體結構,其占據在費米能級處的t*2g簡并軌道在離子嵌入過程中將發生進一步的能級分裂,導致在離子嵌入電化學過程中體系費米能級變化不可控,由此無法定量預測電極電壓的變化幅度。

圖5 (a)橄欖石磷酸鹽LiMPO4和硅酸鹽LiMSiO4(M=Mn、Fe、Co和Ni)中陽離子電負性和電壓的關系[50];(b)八面體配位中M2+陽離子的晶體場分裂。與Mn化合物相比,Fe化合物相對于Li/Li+的電壓較低是由t2g軌道中第6個電子發生配對導致[54];(c)電池電壓由Fe2+/3+的氧化還原能所控制,其由氧化物Fe2O3到聚陰離子氧化物Fe2(MoO4)3,再到具有更強電負性的聚陰離子氧化物Fe2(SO4)3逐漸增加;(d)由分子軌道能級圖可知,Fe2(SO4)3中的Fe2+/3+氧化還原能與同構的Fe2(MoO4)3相比更低,這是由于S-O鍵相較于Mo-O鍵產生的誘導效應對Fe-O共價性的削弱程度更大[59];(e)電池開路電壓示意圖。常見鋰離子電池正極材料和p型摻雜策略調控獲得的LiBCF2/LiB2C2F2電極的電壓、容量與能量密度對比Fig.5 (a)Relationship of cations electronegativity and voltage for olivine phosphates LiMPO4 and silicates LiMSiO4(M=Mn,Fe,Co and Ni);(b)Crystal field splitting of M2+cations in octahedral coordination.Fe compound delivers a lower voltage vs.Li/Li+compared with Mn compound,because Fe2+/3+redox energy shifts due to pairing energy of 6th electron in t2g orbital;(c)Lowering of redox energies of Fe2+/3+couple and consequent increase in cell voltage on going from a simple oxide Fe2O3 to a polyanion oxide Fe2(MoO4)3and then to another polyanion oxide Fe2(MoO4)3 with a more electronegative counter-cation;(d)Molecular orbital energy diagram illustrating lowering of Fe2+/3+redox energy in Fe2(SO4)3 compared to that in isostructural Fe2(MoO4)3,due to a weakening of the Fe—O covalence by a more covalent S—O bond than Mo—O bond through inductive effect;(e)Schematic open-circuit voltage(Voc)of battery.Comparation of voltage,capacity,and energy density of common lithium-ion battery cathode materials and LiBCF2/LiB2C2F2 cathodes obtained by p-type dopingstrategy regulation
值得注意的是,還存在另一類離子嵌入電化學電子結構圖像,即離子嵌入剛性帶模型體系(rigidband model systems)當中,此時離子嵌入帶來的電子將直接轉移到材料費米能級以上的空態而不改變EF附近的分子軌道雜化狀態。因此,可以利用嵌入離子帶來的電子轉移到原始材料費米能級以上的空態所獲得的額外能量預測電極材料電壓,并可能為定量調控電極電壓提供理論指導[62]。基于此在二次電池領域中提出針對剛性帶電極體系(即放電過程中嵌入離子的s電子直接填充至體系導帶底)電子態的p型摻雜策略用于調控上述電極電壓。需要強調的是,p 型摻雜策略通常用于半導體物理領域以調控體系中載流子遷移、電子/空穴對復合等特性,我們則首次呈現其用于離子嵌入電化學過程中通過在電極價帶頂飽和電子處引入空穴實現嵌入離子s電子填充能級的直接調控。據此,通過引入硼原子替換碳原子實現CF體系p型摻雜,并利用CALYPSO結構搜索軟件成功獲得穩定的充電態BCF2/B2C2F2摻雜結構,進一步計算發現其Li+/Na+離子嵌入電壓由2.29 V(CF)提升至3.49/3.63 V(LiBCF2/LiB2C2F2)和2.78/2.85 V(NaBCF2/NaB2C2F2),如圖5(e)所示。由此可見,該p型摻雜策略可廣泛應用于具有剛性帶電荷轉移特性的離子脫/嵌材料體系,為設計新型高比能量正極開辟了新思路[63]。值得注意的是,除上述工作中將p 型摻雜策略應用于不含d 軌道的正極設計中以外,我們將其結合配位場理論定量預測其對離子嵌入電壓的提升作用,還成功提出了一系列新型MS2(M=V,Cr)材料體系,打破了現有提出的電壓最高的MX2電極(TiS2,平均電壓為2.41 V)電壓平臺的突破[64]。本質上這兩個工作都是基于分子軌道理論或者配位場理論對包含sp 電子不同雜化對稱性的體系,或包含d電子在不同配位場下劈裂對稱性的體系進行p型電子態摻雜調控并最終獲得高離子嵌入能量密度正極體系。值得注意的是,上述思想也可推廣至有機電極體系中發生的非離子嵌入式電化學反應的調控,此時鋰離子吸附產生的氧化還原電位依然由體系最高占據分子軌道(HOMO,可相較于無機電極體系的價帶頂)和最低未占據分子軌道(LUMO,可相較于無機電極體系的導帶底)的能級位置決定,并受分子軌道對稱性的嚴格控制[65]。就此,陳軍院士團隊研究了電極還原電位和電子親和(EA)的關聯,發現其呈現出明確的線性關聯。導致這一現象的根本來源在于分子的電子親和能反映了中性分子接受電子而變成陰離子分子的還原過程中自由能的變化,并最終決定了體系放電電壓[66-68]。隨后,Lee等[69-70]也指出有機分子極性和分子大小的不同可能引起較大的溶劑化能差異,并使得電極還原電位和EA 線性關系誤差增大。因此,進一步深入研究有機電極體系中分子軌道對稱性導致的雜化類型與極性等性質的不同,對于調控體系中與LUMO相關特性(如電子親和能等),并提升其電壓性能至關重要。
2.2 電極相結構穩定性
應用于單價/多價金屬離子二次電池的電極材料在離子嵌入/脫出過程中需保持其相結構的穩定性,否則將會導致電池循環/倍率性能的迅速衰減,甚至帶來安全隱患,因此,在設計/調控電極材料電化學性能同時,還需考慮到電極結構的穩定性。眾所周知,在電極材料不同晶格結構的靜電場當中,TM(d)軌道能級將發生不同晶體場劈裂,由此,在電極材料成分、結構設計或調控過程中,特定晶體場劈裂電子構型的能量變化可能會引起電極材料的相變[71-73]。例如,常見MoX2(X=S, Se)電極在離子嵌入過程中H 相(局域結構為三角棱柱場)向T 相(局域結構為八面體場)轉變的機制可以通過配位場理論進行揭示,即H 相MoX2費米能級處電子態主要由飽和Mo-dz2軌道貢獻,由此離子嵌入所帶來的電子將占據更高能級的dxy和dx2-y2)軌道,從而導致H 相結構的不穩定,如圖6(a)所示。相比之下,T 相MoX2中費米能級處的t2g軌道未飽和,由此更易于接受離子嵌入所帶來的電子。這也是導致MoS2在嵌鋰過程中由H 相向T 相轉變的本質原因[74-76]。此外,當離子脫嵌時內部電荷密度將發生顯著變化,如K2Fe(C2O4)2材料中三棱柱FeO6會發生扭曲并轉變為八面體三角反棱柱,由此配體電場對中心Fe原子的dxy和dx2-y2軌道的排斥會增加,導致e''*軌道升高并轉換eg軌道[77-79],如圖6(b)所示。綜上,有必要從配位場理論角度深入探討影響材料結構穩定性的本質。

圖6 (a)電子注入MoX2(X=S,Se)電極材料誘導其由H相向T相結構轉變[74];(b)不同電極相結構中的FeO6分子軌道和其充電/放電狀態下的電子躍遷[77];(c)四面體相(rose)和八面體相(blue)之間的相對穩定性,定義為ΔE[r--b]=E[rose]-E[blue][84];(d)四面體(Td)和八面體(Oh)場的晶體場穩定化能,以及它們之間的差異(以Δ0為單位)[84]Fig.6 (a)Structural phase transition from H-to-T phase induced by injecting electrons into MoX2(X=S,Se)cathodes;(b)Molecular orbitals of FeO6 in different geometries and electron transitions in charged/discharged state;(c)Relative stability between tetrahedral phase(rose)and octahedral phase(blue)is defined as ΔE[r--b]=E[rose]-E[blue];(d)Crystal field stabilization energy of tetrahedral(Td)and octahedral(Oh)fields,and their difference in the unit of Δ0
2.2.1 相結構穩定性與晶體場穩定化能(CFSE)之間的關系
事實上,材料相結構的穩定性與晶體場之間的關聯早已得到系統的解釋。晶體場分裂對系統自由能的影響可以通過以下公式來評估

其中GCFS是晶體場分裂所引起的自由能的變化。在此基礎上Syono 等[80]于1971 年便首次提出應用晶體場穩定化能(CFSE)[81]描述Mg2SiO4-Fe2SiO4材料由橄欖石結構向尖晶石結構的轉變,并提出下列線性關系

其中ΔGCFS、ΔE、ΔV和ΔSCFS分別是尖晶石和橄欖石之間的自由能、內能、體積和電子構型熵差異。根據晶體場理論,d 軌道能級分裂的本質驅動力為晶格能量的降低,其不僅主導了CFSE,而且還可通過消除軌道簡并降低體系的電子構型熵SCFS[82]。由此通過式(8)、式(9),可以將材料不同相結構內能的變化與其CFSE 變化相聯系以判斷相穩定性。
值得注意的是,離子嵌入電化學領域中已有大量研究通過CFSE 來判斷材料不同相的結構穩定性[83]。例如,Manthiram教授等[73]發現層狀Li0.5MO2(M=Co、Ni、Mn)正極中M 的不同導致其轉變為尖晶石相的趨勢不同,這是因為由層狀結構向尖晶石結構的轉變涉及Mn+從過渡金屬層的八面體位點通過附近的四面體位點遷移到鋰層的八面體位點的難易程度。因此,Mn+在八面體與四面體位點中的相對穩定性可能對尖晶石相形成的難易程度起關鍵的作用。通過計算Mn+在八面體位點和四面體位點的晶體場穩定化能的差值,他們發現隨著Mn3+→Ni3+→Co3+八面體位點的穩定性逐漸增加,從而使得Co3+難以向四面體位點遷移,阻礙其向尖晶石狀相Li0.5CoO2的相變。反之,具有較小八面體位點和四面體位點CFSE差值的Li0.5MnO2電極將更易發生向尖晶石相相變。上述結果也與實驗觀察相一致。在此基礎上,Kim等[84]對所有3d過渡金屬處于八面體位和四面體位之間的相對穩定性進行了系統的評估,通過分析兩相之間的能量差異發現,元素周期表左側的3d 過渡金屬更傾向于占據八面體位點,而右側的金屬則總體更傾向于占據四面體位點。上述現象可通過CFSE進行解釋,即不同過渡金屬占據位點的喜好與其所處晶體環境導致其3d 電子的穩定性相關[85]。此外值得注意的是,上述工作中通過將八面體和四面體位點之間的CFSE差值和總能量差異對比還發現,兩者的變化趨勢略有不同,如圖6(c)、(d)所示。這是因為晶體結構中的離子能量與自由態離子能量之差為

其中n1、n2、n3代表軌道的分裂系數,通過查詢表S3可得;Nx、Ny、Nz分別是不同對稱性的d軌道電子數[86];Dq 是晶體場強度,定義為Dq=ZLe2〈r4〉/6R5,〈r4〉是d電子和原子核之間的徑向距離4次方的平均值,R是中心原子和配體之間的平均鍵長,ZL是有效配體電荷;δ和σ都是表示結構畸變的函數,2(δ+σ)/3表示由于結構畸變而獲得的額外穩定焓[87,88],n是成對電子的數量,P代表配對配位能[89]。對于配位能和畸變能的考慮見支撐材料S3部分。由此可見,上述工作中對于CFSE的計算均是基于固定的分裂能(Δ0)來進行的,而實際上Δ0與中心原子和配位場環境密切有關,而非常數,此外電子在不配位場中排布導致的結構畸變能和配位能也不能忽略,因此要得到準確的晶體場穩定化能還需注意對上述因素做進一步的探討。
2.2.2 改進的CFSE模型計算含不同周期元素電極相穩定性
對于包含同一周期過渡金屬元素且其配位環境相同的電極材料相穩定性的比較而言,雖然它們的〈r4〉尚不能準確估計,但是由于其包含的價電子殼層相同,它們的值通常被假定為近似恒定[90]。但是這種簡單的處理方法由于忽略了中心離子本身半徑等屬性的變化,導致可能無法定量反映晶體場分裂與配位場環境之間的關系[64]。對此,Shi 等[91]進一步研究了鹵化物晶體中Ce3+和Eu2+的晶體場分裂與配位場環境之間的定量關系,提出對于同一周期中心原子的〈rk〉可用離子半徑rik進行代替,由此計算出的同一周期過渡金屬中心原子的分裂程度更符合實際情況。
然而,對于包含不同周期過渡金屬元素的材料體系,其d軌道本身的特性不同,例如電子殼層和核電荷數都有很大的差別,導致Dq中〈r4〉的數值將發生顯著改變,因此無法簡單采用rik4代替〈r4〉定量獲得晶體場分裂程度的大小,此時必須重新從體系哈密頓量出發進行探討。理論上金屬離子的哈密頓量通常寫成自由金屬離子和包含配位場參數Akq的單電子之和,因此該模型中哈密頓量可以寫成

其中〈rk〉是中心離子的徑向積分,Akq是僅取決于配位環境的晶體場參數,與中心離子無關。Cqk是電子的坐標參數,可以根據電子的角坐標計算。一般認為,Dq 可寫成配體函數f和中心離子g的乘積:Dq=g(中心離子)×f(配體)=ZLe2〈r4〉/6R5[92],可見中心原子周期不同,徑向分布函數明顯不同。為了定量計算出中心原子d軌道的分裂程度,首先考慮宿主材料晶體場環境的影響,可采用離子半徑ri4來代替〈r4〉,因此Dq 可簡化為Dq=ZLe2ri4/6R5;其次考慮對具有不同d 軌道殼層的中心離子進行修正,即Dqgf=m×ZLe2ri4/6R5,此時可將具有相同配體的中心離子劃定為同一個光譜化學系列,并根據過渡金屬基團價電子殼層數的增加(3dn∶4dn∶5dn)或者價態變化(+2∶+3∶+4)定義m函數的相對值比例分別為1∶1.45∶1.75和1∶1.6∶1.9[92]。由此,上述定義的晶體場劈裂強度Dqgf=g×ZLe2ri4/6R5可用于計算不同周期過渡金屬d軌道能級分裂情況,并據此考察含上述不同周期過渡金屬體系不同相結構(α→β)的相對穩定化能(ΔCFSEα→β)。
根據上述定義的Dqgf,分析了含不同周期(5d→3d)過渡金屬的MX2(X=S, Se)體系2H 相和1T 相結構相對穩定性和CFSE的關系,如圖7 所示。首先根據第一性原理計算結果可以看出[圖7(a)],隨著過渡金屬中心原子周期的減少(W→Mo→Cr),MX2體系2H和1T相之間的能量差(ΔE1T-2H)逐漸減小。隨后通過計算VIB 族MX2(M=Cr, Mo, W;X=S,Se)體系不同相結構晶體場穩定化能之差[ΔCFSE1T-2H=CFSE1T-CFSE2H,其中CFSE1T=-8×Dq1T,CFSE2H=-11.68×Dq2H,圖7(b)]可知,ΔCFSE1T-2H始終為正值,說明2H 相始終為最穩定相。最重要的是,隨著MX2(M=Cr, Mo, W;X=S,Se)中陽離子周期的減小,ΔCFSE1T-2H逐漸減小,驗證了上述第一性原理計算發現系統有從2H相向1T相轉變的趨勢。由此可見,電極材料在調控過程中可能存在的相變可以由晶體場理論推導d軌道分裂情況直接預測,因此,通過CFSE衡量材料體系的相結構穩定性為進一步同步調控材料電壓和穩定性等多性能提供新思路。最后值得注意的是,上述預測MX2(M=Cr,Mo,W;X=S,Se)的ΔCFSE1T-2H和第一性原理計算得到的ΔE1T-2H在Mo 處的拐點不一致是由于晶體場理論預測中忽略了結構畸變等非理想情況因素的影響,如式(10)中討論,這也為進一步改進上述晶體場穩定化能預測精度提供方向。

圖7 (a)VIB族MX2(M=Cr,Mo,W;X=S,Se)體系1T相和2H相之間的結構穩定性:ΔE1T-2H=E[1T]-E[2H];(b)MX2電極相變前(2H)和相變后(1T)的晶體場穩定化能之差(ΔCFSE1T-2H),橙色和藍色標簽分別代表Se族和S族化合物Fig.7 (a)Relative stabilities between 1T and 2H phases of MX2(M=Cr,Mo,W;X=S,Se)is defined as ΔE1T-2H=E[1T]-E[2H];(b)Differences of MX2 crystal field stabilization energies before(2H)and after(1T)phase transitions.Orange and blue labels represent Se and S compound,respectively
2.3 電極陰離子氧化還原活性
由于富鋰氧化物在充放電過程中存在涉及陽離子和陰離子的多中心氧化還原過程,具有超高的比容量,因而被認為是目前最有前途的高能量密度正極候選體系[93-94]。先前的理論研究表明,富鋰材料中的陰離子氧化還原活性可歸因于富鋰氧化物中普遍存在的特殊Li-O-Li 局部構型所誘導產生的氧2p非鍵態(NB)[95-96],如圖8(a)所示。隨后一系列后續研究表明,氧2p非鍵態不僅在Li-O-Li構型中產生,而且在特定軸上存在不與氧2p 軌道形成雜化的離子如Na+、Mg2+甚至過渡金屬空位時也同樣產生。然而,陰離子參與變價往往伴隨著循環穩定性差、電壓衰減,并且可能還會導致嚴重的安全問題,這些限制因素成為富鋰材料在實際應用中使用的瓶頸,促使研究人員通過進一步理解陰離子氧化還原行為提出改進的策略[95,97]。
2.3.1 陰離子氧化還原電化學的起源

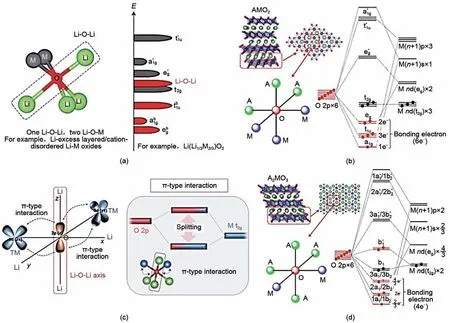
圖8 (a)富鋰層狀氧化物(如Li2MnO3)中Li-O-Li構型的局部原子配位及其能帶結構示意圖[96];(b)層狀過渡金屬氧化物AMO2中OA3M3八面體的分子軌道能級圖[106];(c)Li-O-Li電子和M-t2g被視為π型雜化態情況下的能帶結構示意圖[107];(d)富A層狀氧化物A2MO3中OA4M2八面體的分子軌道能量圖[106]Fig.8 (a)Local atomic coordination around oxygen with one Li-O-Li configurations in Li-rich layered oxides(such as Li2MnO3)and schematic of their band structures;(b)Molecular orbital energy diagrams of an OA3M3 octahedron in layered transition-metal oxides AMO2;(c)Schematic illustration of band structure for-case where-Li-O-Li axis and M-t2g are regarded as a π-type hybridized state;(d)Molecular orbital energy diagrams of an OA4M2octahedron in A-rich layered transition-metal oxides A2MO3
由此可見,對于傳統的AMO2電極材料,其容量基本上來自陽離子的氧化還原反應,即使在M上產生的空穴通過軌道雜化在一定程度上離域到O-2p 能級上[99-100],但由于所有O-2p 電子都具有很強的成鍵特性并占據到能量更低的能帶,阻礙了額外的氧的氧化,從而無法產生任何的額外容量[101-102]。對于富鋰材料,當氧由2 個M 和4 個A 配位時,OM2A4八面體的點對稱性為C2v。就此,雜化分子軌道可以重新標記如下:4 根O-2p 軌道與M-nd(eg),(n+1)s和(n+1)p軌道形成σ型成鍵(a1和b2)和反鍵態(a*1和b*2)分子軌道;形成Li-O-Li 構型時,兩個O-2p 軌道沿A-O-A 軸和兩個M-nd(t2g)軌道形成π 型成鍵(b1)和反鍵(b*1)分子軌道[95,97,103-104],如圖8(c)所示。在典型陰離子氧化還原電極中的高價M離子(Mn4+,Co4+,Ni4+,Ru5+,Ir5+),M-nd(t2g)軌道的能級低于O-2p的能級。因此,占據的b*1分子軌道主要由O-2p組成,主導氧的氧化還原反應,如圖8(d)所示。該圖像進一步綜合考慮了σ 型和π型雜化,但是一些非鍵態作用未考慮在內。更重要的是,在氧的氧化還原反應發生之前,O-2p 和M-nd(t2g)之間的這種π 型相互作用很弱,并且費米能級附近的O-2p軌道似乎是“孤立的”[105]。然而,在反鍵b*1分子軌道發生氧化后,相應的M-O鍵成鍵效應變得更加緊密,并導致更大的b1/b*1分裂,從而穩定被氧化的晶格氧原子并產生額外的容量。
值得注意的是,通常認為的Li-O-Li 構型是孤立態,其能級與過渡金屬離子無關的觀點,與富鋰材料中氧的氧化還原活性與過渡金屬元素密切相關的事實矛盾。事實上過渡金屬種類會影響陰離子氧化還原活性。例如,對于同周期過渡金屬元素,Li2TiO3中的陰離子氧化還原在一般電化學窗口中不提供任何實際容量[108],而具有相同層狀結構的Li2MnO3中的陰離子氧化可以提供顯著的高充電容量,這說明同周期過渡金屬的類型會影響陰離子氧化還原活性[109]。類似地,對于不同周期的過渡金屬類型,在Li1.2Ni0.2M0.6O2(M=Mn,Ru)化合物的比較研究中,僅Li1.2Ni0.2Mn0.6O2發生氧的氧化還原,而Li1.2Ni0.2Ru0.6O2的陰離子氧化還原活性可以忽略不計,這說明Mn和Ru的價電子性質會影響陰離子氧化還原活性[110]。上述結果表明Li-O-Li構型和過渡金屬之間存在緊密的相互作用,因此有必要利用考慮了原子軌道分裂狀態和雜化分子軌道能級對稱性耦合的配位場理論對上述陰離子氧化還原機制進行探討。
2.3.2 陰離子氧化還原行為調控研究進展
對于過渡金屬種類對氧氧化還原活性的影響,Song等[107]以3d過渡金屬基Li2MO3化合物為例,分析受過渡金屬中心原子種類影響的氧的局部環境與O-2p 氧化還原活性之間的相關性,為各種Li2MO3化合物中氧離子氧化還原的惰性/活性提供了合理的解釋。通過考慮本征O-2p 和M-t2g軌道的相對能級,可以闡明π型雜化帶的排列。圖9中給出了兩種可能的情況下O-2p和M-t2g形成π型雜化示意圖,π型雜化的成鍵和反鍵態能級分別表示為b1和b*1[106]。對于第1 種情況(圖中Case 1,即O-2p<M-t2g),b1能級主要由氧離子控制,而金屬特性在b*1水平上占主導地位。而對于第1 種情況(圖中Case 2,即O-2p>M-t2g)則狀態相反。

圖9 根據O-2p和M-t2g的相對位置推測可形成的能帶結構示意圖,紅色和藍色分別表示氧和金屬特性,橙色陰影區域是Li6-O態的能級,即未雜化的O-2p能級[107]Fig.9 Schematic diagram of band structure that can be formed depending on relative positions of O-2p and M-t2g.Red and blue colors indicate oxygen and metal character,respectively.Orange shaded region is energy level of Li6-O state,unhybridized O 2p level
根據晶體場理論,M-t2g的相對能級高低取決于晶體場分裂能的相對大小。由于Li2MO3化合物局部環境是八面體結構,較大的分裂能(Δ0)[111]會導致Mt2g能級較低。八面體場中的M-3d 軌道分為高能級eg軌道和低能級t2g軌道。eg和t2g軌道之間的能量差為Δ0=E(eg)-E(t2g)=(+6×Dq)-(-4×Dq)=10×Dq=5ZLe2〈r4〉/3R5,其中ZL是配體有效電荷,e是電子電荷,R是過渡金屬和配體之間的平均原子距離,r是電子到原子核的徑向距離,〈r4〉是電子和原子核之間的徑向距離4次方的平均。對于同一過渡周期系列中具有相似價態數的陽離子,〈r4〉的值被假定為近似恒定,這表明3d 過渡金屬基Li2MO3化合物的Δ0主要通過M-O 的平均原子距離(R)來區分,3d 過渡金屬基Li2MO3化合物隨著原子序數的增加,M和O 之間的平均原子間距離逐漸減小使得Δ0單調增加,從而導致M-t2g能級較低。這種趨勢也類似于常規正極中過渡金屬的一般電壓趨勢,其中M-t2g被認為是未雜化狀態[112-113]。
考慮到Δ0隨著M的原子序數單調增加,對于M原子序數較小的Li2MO3化合物一般屬于圖9 中第1 種情況(O-2p<M-t2g),而第二種情況(O-2p>M-t2g)在M 原子序數較大的Li2MO3化合物中占據主導地位。結合TDOS圖和配位場理論分析,可以定量分析M-t2g和O-2p 能級的相對位置對陰離子氧化還原活性的影響。考慮到從元素周期表前期到后期的Δ0逐漸增加,后期Li2MO3化合物的M-t2g能級相對O-2p能級逐漸降低。圖10說明了這些b1和b*1狀態如何從Ti 到Ni 的過渡金屬系統的變化特征。可以看到,價帶最大值的特征由填充在π型雜化狀態中的電子數量決定,且過渡金屬的不同導致了陽離子和陰離子變價性質的本質變化[113-115]。上述工作證實了利用配位場理論對M-t2g軌道能級中的電子數和M-t2g軌道能級與O-2p 能級的相對能級位置進行分析可直接幫助指導富鋰體系中陰離子氧化還原活性的調控。

圖10 層狀Li2MO3(M=Ti,V,Cr,Mn,Fe,Co,Ni)電極的電子能帶結構示意圖。分子軌道的數量按是按單個氧原子為標準給出的。黑色實心圓圈代表電子,不同圓圈缺口取決于它們貢獻電子數的多少。每個狀態上的紅色、藍色和黃色陰影分別表示氧主導、金屬主導以及金屬和氧競爭的特征[107]Fig.10 Schematic illustration of electronic band structures of honeycomb ordering layered Li2TMO3(TM=Ti,V,Cr,Mn,Fe,Co,Ni)cathodes.Number of each molecular orbital was normalized per oxygen.Black dots represent electrons,and size of dots indicates their contribution.Red,blue,and yellow blurring on each state indicate oxygen dominant,metal dominant,and metal and oxygen competitive character,respectively
在此基礎上,夏定國和艾新平等[116]提出綜合考慮富鋰材料中成鍵/非成鍵π 型軌道、氧非鍵軌道和成鍵/非成鍵σ 型軌道探討O-2p 參與氧化還原的完整圖像。發現Li2RhO3中氧的氧化還原可以通過Rh-t2g與氧之間較強的π 型雜化所形成的反鍵態在低電壓下活化,這種雙峰氧氧化還原行為首次出現,并且是由富鋰材料中氧鍵的復雜性質引起的。上述研究豐富了我們對富鋰材料中氧的氧化還原行為的理解,并為未來高容量富鋰正極材料的設計提供啟示。綜上所述,從配位場理論的角度探討了陰離子氧化還原的理論調控機理,并引發了對陰離子氧化還原電化學背后電子結構圖像更深入的思考。
2.4 過渡金屬離子遷移行為
在過渡金屬基正極的離子嵌入電化學反應過程中,除了備受關注并決定電池倍率性能的單價/多價金屬離子遷移外,往往還伴隨著由氧化還原反應驅動的過渡金屬離子遷移,如層狀二元或三元過渡金屬正極材料中金屬離子(TMn+)從TM 層遷移至單價/多價金屬層[117-118]。然而,這種TMn+的遷移行為甚至發生TM層/遷移離子層互占位現象可以促進新的電化學變價機理的產生[119-121],也可能會阻礙單價/多價金屬離子的擴散,并引起材料在可逆容量、首次效率、倍率性能等方面電化學性能的下降。
2.4.1 過渡金屬離子遷移行為
過渡金屬離子遷移現象最初是在LiNiO2材料離子脫出過程中提出。發現體系在鋰離子脫嵌過程中Ni2+將從TM層遷移到堿金屬層導致LiNiO2的結構紊亂并影響材料的電化學性能[122-125]。當在鋰離子層中Ni 與Li 有50%的反占位時,層狀的LiNiO2轉變為無序巖鹽結構,大大削弱鋰離子的電化學活性。TM 離子遷移現象不僅在傳統金屬氧化物中存在,在富鋰材料中也備受關注。由于材料的可逆容量是通過鋰離子在正極晶格中可逆地嵌入/脫出實現的,但是TM 離子遷移會造成鋰離子可逆脫嵌的數量減少,降低了材料的可逆容量[126-129]。因此,研究TM離子遷移發生的原因以及對材料電化學性能的影響至關重要。
研究人員通常是從電極材料在離子脫嵌過程中的微觀結構變化角度入手來揭示過渡金屬離子遷移對電化學性能的影響。王崇敏等[130-131]發現在鋰離子層狀化合物循環過程中TM 從表面朝體相方向傳播,并且在多次循環后,電極將發生從層狀到無序巖鹽結構的轉變,最終導致在層狀富鋰材料中發現尖晶石結構的出現,如圖11(a)所示。正是由于上述不可逆相變的發生,導致在鋰離子電池電化學循環性能的急劇衰減[130,132-133]。此外,有研究表明過渡金屬離子遷移行為不僅發生在鋰離子電池中,也同樣發生在鈉離子電池中[117,134]。在α-NaFeO2、NaCrO2和NaCoO2等電極材料深度脫鈉狀態下,TM 離子從TM 層遷移到Na 層通常會導致Na+傳輸受阻和TM 層被破壞,導致電極容量的急劇衰減。為了進一步理解上述衰減機制,Kubota 等[135]通過研究NaCrO2發現,脫Na過程中由于Cr離子從CrO2的八面體位遷移至Na層間的四面體和八面體位,導致材料發生P'3 相到O3'相的不可逆轉變,從而嚴重影響著材料的電化學性能,如圖11(b)所示。

圖11 (a)NMC333正極表層的STEM-HAADF圖像展示從表面到體積的轉換的結構模型,其中過渡金屬離子、鋰離子和氧離子分別用藍色、綠色和紅色球表示。黃色虛線表示表面重構層。圖中3個不同區域的快速傅里葉變換顯示在底部,并呈現出結構的變換。藍色箭頭表示額外顯現的斑點,黑色虛線框突出顯示由于晶格紊亂引起的晶胞變化[130];(b)Cr在NaCrO2的遷移過程[135]Fig.11 (a)STEM-HAADF image of positive electrode surface layer of NMC333 shows structural model of lattice transformation from surface into bulk,where transition metal ions,Li ions,and oxygen ions are denoted by blue,green,and red balls,respectively.Yellow dashed lines denote surface reconstruction layer.Fast Fourier transformation from 3 different regions in panel d are shown at bottom,indicating structure transformation.Blue arrows indicate extra spots,and dashed black frames highlight unit cell changes due to lattice disordering;(b)Migration process of Cr in NaCrO2
因此,抑制TM 離子遷移的行為可能是改善這類電極電化學性能的有效方法之一。眾所周知,在常見的Li2TMO3(TM=過渡金屬)電極中,TM離子可以從TM 層的八面體位置遷移到Li 層的四面體位置,然后進一步遷移到Li層的八面體位置。摻雜多價陽離子是直接抑制TM 離子遷移的重要手段,外來多價陽離子由于其較大的原子半徑可以阻斷TM遷移路徑或者通過與氧形成更強的鍵來增強結構穩定性。由于硼半徑小,它可以插入TM 層的四面體位置,阻礙TM 遷移的路徑,從而阻止不可逆相變的發生,如圖12(a)、(b)所示。強B-O鍵可以增強摻硼富鋰材料(LLTO@LBO)的結構穩定性,且硼原子具有較輕的原子質量,可最大程度上減少非活性元素的引入導致的容量衰減,由此使得摻雜的LLO@LBO電極具有優異的電化學性能[136]。在鈉離子電池中,P2 型Fe 基材料由于Fe3+遷移到鄰近的四面體位置,導致不可逆相變的發生,造成了容量的損失。Cu2+/Cu3+氧化還原對在鈉離子電池中表現出電化學活性,將過量的Cu 摻雜到晶格中,可以大大減輕Fe 的遷移,因此加強了結構的穩定性。此外,Cu 的引入增強了過渡金屬元素和配體氧之間的電子耦合強度從而帶來了額外的容量,這可以

圖12 (a)LLO@LBO中Mn的遷移模型[136];(b)Mn在LLO@LBO和LLO中不同的遷移能壘[136];(c-d)O3型和O2型鋰層狀氧化物中TM遷移路徑的示意圖。紅色和黃色箭頭分別表示層間和層內TM遷移。黑色箭頭表示Li位點中的TM離子與TM層中的TM離子之間的靜電斥力。O3結構中的TM離子較易占據與相鄰陽離子共邊的Li位點,然而O2結構中的TM離子占據Li位點則會受到強烈的排斥力[138]Fig.12 (a)Migration module of Mn in LLO@LBO;(b)Energy of Mn at different positions in LLO@LBO and LLO;(c-d)Schematic illustrations of crystal structures of O3-type(c)and O2-type(d)lithium layered oxides TM migration paths.Red and yellow arrows indicate interlayer and intra-layer TM migrations,respectively.And black arrows represent electrostatic repulsion between a TM ion in Li sites and TM ions in TM layer.Although TM ions in O3 structure can readily occupy Li sites that share only edges with neighboring cations,TM ions in O2 structure are subject to strong repulsion when they occupy Li sites face-sharing with neighbouring cations
需要注意的是,并非所有TM 離子遷移都是不可逆的。Eum 等[138]通過合成O2 結構的Lix(Li0.2Ni0.2Mn0.6)(x<0.830),發現在兩種結構中TM離子都從TM 層的八面體位置遷移到堿金屬層的四面體位置。但是上述兩種相結構最大的區別在于O3結構中的TM 會在堿金屬層進一步遷移,但在O2相中TM 進一步向堿金屬層遷移的勢壘較高,因此在放電的過程時將可逆遷移回TM 層,大大地提高了O2 結構材料的充放電循環穩定性,如圖12(c)、(d)所示。此外,TM遷移還可能為離子嵌入電化學領域帶來新的氧化還原反應機制。我們早期的研究中提出在層狀NaCrS2材料中新型單陰離子氧化還原電化學的存在,這種新的S22-/S2-單陰離子氧化還原活性是由脫鈉過程中鉻離子遷移到鈉層形成鉻/鈉空位反位(Cr/V'Na)’導致[119]。因此,從配位場理論角度出發,研究不同電極材料中TM 的可逆遷移機制可以為提高富鋰正極的循環穩定性或探索新型氧化還原活性機理提供理論指導。
2.4.2 配位場下TM離子的遷移行為
揭示TM 離子的遷移對調節電極相的穩定性以及理解陰離子氧化還原機制起著關鍵性的作用。然而,帶有離域d 電子的TM 受到配位場的控制,其遷移行為比僅包含s價電子的堿金屬或堿土金屬離子的遷移行為更為復雜。例如,在離子嵌入電化學過程中過渡金屬電子排布的變化可能會導致Jahn-Teller 畸變的產生,從而對離子遷移產生重要影響[139]。因此,下述將從晶體場理論出發,為研究材料中本征或摻雜的過渡金屬的遷移性質帶來獨特且全面的理解。
事實上,TM 離子的遷移勢壘取決于其潛在遷移路徑上初態-過渡態-終態的能量差。如圖13(a)很好地補償惰性雜質摻雜造成的容量損失[137]。中所示,在典型的層狀正極材料(LiNiO2、LiMnO2等)中TM 離子遷移路徑為八面體-四面體-八面體。而從八面體位遷移到四面體位時,TM-d 電子軌道能級將從t2g(-4Dq)和eg(+6Dq)改變為e(-2.67Dq)和t2(+1.73Dq),這也導致了TM 占位能量的改變。由此可以看出,研究TM 離子遷移必須考慮到配位場變化產生的能量差異。首先,可根據電極中含不同價電子的TM 在不同配位場中電子占據的特征,分析得到其占位偏好,即TM 占據的基態位點。例如,對于Mn4+而言,其通常將穩定占據在八面體位點,而Mn5+則以四面體占位為主。導致上述現象的本質是由于八面體中t2g和eg軌道的分裂程度強于四面體中e 和t2的分裂,因此,從Mn4+的低能級t2g軌道能級上去除電子以產生Mn5+要比從高位的四面體t2軌道能級去除電子困難得多,由此導致Mn5+更傾向于占據在四面體位置。另外對于Mn4+和Mn3+,其位點偏好也有所不同,Mn4+有3 個d 電子填充在t2g軌道上,導致八面體結構比較穩定[140];對于Mn3+的形成過程中容易發生電荷歧化反應,形成四面體結構。這里需要強調的是,將初態和過渡態之間的能量差進行計算,可以用來評估TM 離子遷移的難易程度,如圖13(b)所示。Ceder 等[141]將3d 周期的TM 離子從八面體位點向四面體位點遷移的能量差進行了計算,發現其能量差主要由d電子數、晶體場強度以及配位能決定。其中,TM 離子的種類以及價態決定了軌道d電子數;而晶體場強度和配位能的討論則可見上述2.2 節論述。由此也可以很好解釋實驗當中觀察到的Fe3+在八面體和四面體之間容易發生離子遷移,這是由于Fe3+在這兩種晶體場中穩定能的差值為0,當脫嵌的過程中產生足夠高的鈉空位時,Fe3+很容易遷移到與八面體共面的四面體位點[142]。而對于Ni4+,八面體和四面體之間的

圖13 (a)層狀正極材料中典型的TM離子遷移路徑為八面體-四面體-八面體;(b)TM離子遷移的難易程度。從位點A遷移至位點B需要消耗額外的能量Fig.13 (a)Typical migration path of TM ions in layered cathode is octahedron-tetrahedron-octahedron;(b)Difficulty of TM ion migration in systems.Migrating from site A to B requires additional energy
綜上所述,探討了TM 離子遷移對材料性能的影響,從配位場角度揭示出TM 離子遷移的本質原因。TM 離子遷移對材料性能的影響以及背后的科學問題更值得深思,如上所述在NaCrS2電極材料中Cr 遷移到Na 層與Na 空位形成反占位后可導致單陰離子變價的產生。由此,我們希望從配位場理論角度出發能夠為探索新型電極材料及新電化學變價機制提供研究的新思路。
3 結論與展望
單價/多價金屬離子二次電池是基于離子在正負電極之間來回穿梭進行電化學反應實現能量的存儲與釋放。通常正極材料由離子嵌入式過渡金屬化合物組成,其電子結構取決于過渡金屬的d軌道性質,并受到晶體配位場的嚴格控制。由此,基于配位場理論建立材料局域環境對離子嵌入過程中的電壓平臺、相結構穩定性和比容量等電化學性能的影響機理至關重要。本文從配位場理論出發,結合第一性原理計算分子軌道能級及電子占據等信息的相互佐證,系統分析了材料中的局域結構屬性決定其電化學性能的本質機理,并推導出了調控關鍵電化學性能的模型/理論,如電極電壓平臺、結構穩定性、比容量和離子遷移等。上述思想將為新型電極材料的設計提供一個新的視角。在此基礎上,我們提出了新型電極材料的設計方向如下。
(1)過去十多年,科研人員對除鋰離子以外的其他電池開展了廣泛的探索,重新激發了人們對具有超高理論比容量(3870 mA·h/g)的鋰金屬負極這一二次電池中“圣杯”的追求。目前隨著固體電解質的出現和鋰枝晶安全問題的改善,使得鋰金屬負極的回歸成為可能。由此,亟待設計出高電壓無鋰嵌入式正極材料與鋰金屬負極相匹配,這也成為突破目前鋰離子電池能量密度限制最有前途的方法之一。設計高電壓無鋰正極材料還需要從2.1 節所述Goodenough教授提出的電子結構圖像出發,將嵌入式電極電壓的改善問題回歸到對體系費米能級的調控這一關鍵問題上來。因此,從配位場理論出發研究費米能級處的雜化分子軌道特性,將為提出調控有效的理論調控電壓策略提供指導。
(2)富鋰正極材料具有容量大、成本低、工作電位高等優點,但是在高壓下通常伴隨著循環穩定相對穩定化能差值較大,導致其難以發生遷移。性差、電壓衰減和相關的嚴重安全問題,這些限制因素已成為富鋰材料在實際應用中使用的瓶頸。材料中的缺陷(氧空位、摻雜等)或相變(堆垛層錯)的發生有利于激活氧活性,提高容量,但也可能會導致性能衰減。因此,如何利用配位場理論來調控過渡金屬與陰離子軌道能級相對位置、電子占據及分布狀態并找到容量穩定性與性能衰減之間的平衡點,也是新型富鋰正極材料設計的重要研究方向。
(3)當前新型電極材料的設計主要聚焦于過渡金屬基電極材料的設計,但基于配位場理論調控非過渡金屬基電極材料,甚至是有機電極也正成為當前發展的主要方向之一。雖然這些非過渡金屬基的電極材料不含有d軌道,但是依然可以在滿足對稱性匹配原則、最大重疊原則和能量相近原則的情況下,采用分子軌道理論來調控上述電極費米能級與電壓之間的關系,因此也是未來進一步提出定量調控電極電壓有效策略的潛在方向之一。
然而,目前配位場理論目前存在的局限性包括。
(1)配位場理論模型是基于點電荷模型提出的,點電荷模型能夠計算d軌道的分裂,但是其分裂程度被嚴重低估。這本質上是由于配位場理論模型僅考慮了中心離子與周圍配位離子之間的庫侖作用而忽略了更多的相互作用,如不同離子的電子結構的差異、配體的性質、電子密度的空間分布、中心離子和配體之間的化學鍵等因素。因此,迫切需要就上述因素對d軌道的分裂進行詳細探討并提出修正模型。
