一種罕見小額外標記染色體的鑒定及臨床意義分析
2022-03-05 08:26:48王紅丹夏海蘭李永樂張曉梅馮戰啟
安徽醫科大學學報 2022年2期
王紅丹,夏海蘭,李永樂,高 越,張曉梅,馮戰啟
小額外標記染色體 (small supernumerary marker chromosomes, sSMC) 是不能夠被傳統的染色體核型分析技術識別的多余的結構異常染色體[1]。大約60%的sSMC衍生于染色體短臂或者近端著絲粒染色體近中心區域。人群中sSMC的發生率為0.6/1000左右[2],是一種罕見染色體異常。25%~30%被發現的sSMC產生于15號染色體,sSMC(15)是最常見的sSMC類型[2]。起源于10號染色體的sSMC更為罕見,截至目前國內外僅見10余例報道。sSMC的臨床效應取決于sSMC中常染色質的含量,染色體的起源以及嵌合比例等因素,因此,為了更好地了解核型與表型的相關性,繼續研究報道更多的sSMC病例,特別是起源于10號染色體的罕見特征性病例。該研究對一攜帶sSMC(10)的患者進行了臨床表型、遺傳特征及臨床意義分析。
1 材料與方法
1.1 病例資料患兒母親,29歲,G3P1,有兩次早孕期胎停史,未進行遺傳咨詢。患兒父親,30歲,既往體健。患兒,女,7月齡,因新生兒窒息、缺血缺氧性腦病、生長發育遲緩來河南省人民醫院醫學遺傳研究所遺傳門診進行遺傳咨詢,患兒體檢未見明顯結構畸形。患兒父母已經簽署知情同意書,本研究通過河南省人民醫院倫理委員會批準。
1.2 方法
1.2.1細胞遺傳學核型分析 取患兒及其父母肝素鈉抗凝血2 ml,接種于人外周血淋巴細胞培養基中,置于37℃恒溫培養箱中,持續培養72 h,收獲前1 h加入秋水仙素,經過氯化鉀低滲處理、固定、滴片和烤片等常規過程制片。胰酶消化后采用Giemsa染色進行顯帶。采用全自動染色體核型分析系統(德國LEICA公司)計數30個分散良好且顯帶清晰的分裂相進行分析。
1.2.2染色體芯片分析(chromosomal microarray analysis, CMA) 采集患兒及父母外周血2 ml,應用柱式全血基因組DNA提取試劑盒 (德國QIAGEN公司)提取基因組DNA。采用Qubit定量平臺(美國Qubit 3.0)定量DNA的濃度。應用Agilent公司生產的 SurePrint G3 Human CGH Microarray 8×60K芯片(美國Agilent公司)對患者及其父母全基因組CNVs進行檢測。實驗原理是將患者樣本DNA與正常對照樣本DNA分別用不同的熒光素標記(cy3/cy5)標記、純化后進行競爭性雜交,采用Agilent Microarray Scanner對芯片進行掃描,運用Agilent CytoGenomics Edition軟件得到定量的拷貝數檢測結果。軟件參數設置為對基因檢測片段中連續3個探針logR值≥0.25或logR≤-0.25的CNVs進行標示。據文獻[3]報道,大約99.34%致病性拷貝數變異(copy number variation, CNVs)長度均大于300 kb,對200 kb以上CNVs進行檢測并做出提示。此芯片檢測不能排除更小的染色體結構異常的可能性。芯片序列信息來源于2009年2月UCSC數據庫提供的人類基因組參考序列(GRCh37/hg19)。
1.2.3影像學檢查 患兒于出生第2天及第11天進行了MRI檢查,序列:IR FRFSE SE/EPI;AXI:TIFLAIR&T2FLAIR,FRFSE T2WI;SE/EPI DWI SAG:T1FLAIR。
2 結果
2.1 細胞遺傳學檢測結果患兒母親染色體核型為46, XX, t (10; 13)(p11.1;q11)[11]/ 46, XX[19](圖1)。患兒父親染色體核型結果正常,核型為46,XY。患兒染色體核型為47, XX, +mar(圖2)。

圖1 患兒母親G顯帶染色體核型分析圖
2.2 CMA技術分析結果患兒父母CMA檢測結果顯示不存在200 kb以上CNVs。患兒CMA檢測結果顯示10號染色體p15.3p11.1區域LogR值顯著升高,大約在0.5附近,提示患兒10號染色體p15.3p11.1區域發生了3拷貝重復。CMA分子核型為:arr[hg19] 10p15.3p11.1(148,206~38,545,928)×3,片段大小為38.39 Mb,13號染色體未見重復和缺失(圖3)。
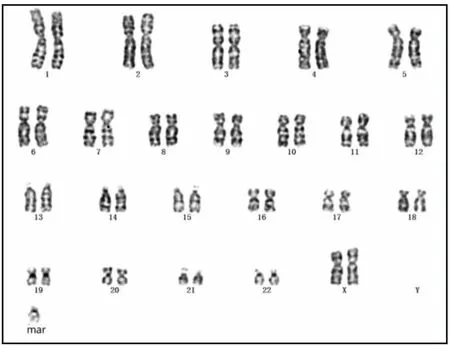
圖2 患兒G顯帶染色體核型分析圖
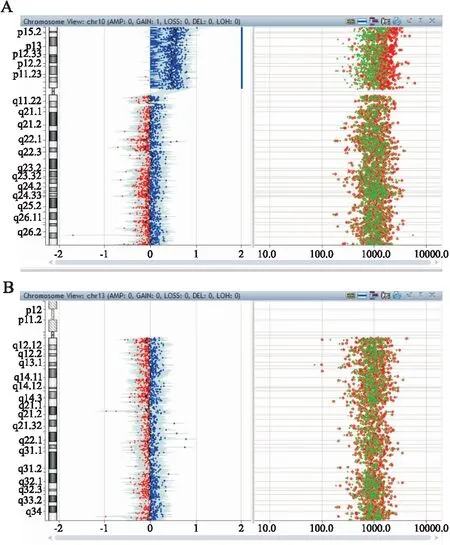
圖3 患兒CMA檢測結果
2.3 影像學檢測結果患兒于出生第2天進行了頭部MRI檢查。可見左側顳葉、右側枕葉異常信號,雙側側腦室后角內積血,符合缺氧缺血性腦病;Dandy-Walker畸形,枕大池內有積血;胼胝體異常信號及左側頭皮下血腫。出生第11天進行了頭部MRI復查,與前片相比,右側額葉新見異常信號區,有局部腦軟化形成趨勢,余同前片。CT檢測結果支持缺血缺氧性腦病。
2.4 回顧性總結以往sSMC(10)病例對文獻報道的sSMC(10)病例進行了遺傳學和臨床總結。見表1。

表1 文獻報道的sSMC(10)病例的遺傳學及臨床特征
3 討論
加用傳統核型分析以外的遺傳學檢測方法,明確sSMC的衍生來源、大小、嵌合比例、組成結構以及包含的基因等顯得尤為重要,是臨床進行遺傳學診斷及產前診斷的依據。針對有明確臨床表型,同時核型檢測到sSMC的病例,目前臨床多采用CMA進行復查及確診。但是需要注意的是CMA不能檢測到平衡易位、倒位、低比例嵌合及多倍體等情況。
各個報道給出的sSMC攜帶頻率不盡相同,目前尚無中國人群攜帶sSMC頻率等相關報道。據文獻報道,sSMC在人群中比較罕見,本病例代表一種新的sSMC(10)細胞遺傳學形成機制,這在以往尚未見報道。課題組應用CMA技術對患兒基因組DNA進行分析,結果顯示CMA分子核型為:arr[hg19]10p15.3p11.1(148,206~38,545,928)×3,片段大小為38.39 Mb。結合父母核型結果,分析患兒mar可能主要是由10號染色體短臂組成。
由于sSMC的常染色體組成、與sSMC同源的親本染色體嵌合比例以及單親二倍體程度等的不同,特定sSMC的表型是難以預測的。研究者普遍認為,攜帶sSMC的表型主要取決于sSMC中常染色質的含量以及嵌合比例。來自于10號染色體的sSMC非常罕見,本實驗對已報道的sSMC(10)案例進行了總結,詳見表1。這些病例中,其中有5例出現異常表型。未見異常表型的染色體來源區段分布在10p11.2q11.2以及10pterp14-15[6,11-12]。Trimbom et al[10]報道的患者是一個例外,報道了一個攜帶環狀sSMC的14歲女孩(淋巴細胞嵌合比例為14%)呈現出生長發育遲緩、智力低下、肌張力減退、注意力下降等表型。由于技術手段的局限性,這些病例并未進行單親二倍體檢測,如果sSMC同源的親本染色體來源于一個親本,這樣的患者是有可能表現出異常表型的。排除單親二倍體以及其它未檢測到的遺傳學改變的情況下,臨床上遇到10p11.2q11.2的sSMC,可以認定為良性變異。Snyder et al[5]和Chen et al[8]報道的病例幾乎涉及到整個10號染色體短臂,患者出現了腎臟發育不全,指甲發育不全等多發畸形。Blennow et al[4]報道的10p11.1p12是涉及范圍最小的致病性sSMC,本病例顯示10p11.2p12可能是比較重要的致病區域,但其對sSMC范圍的認定可能不是那么精確,且未進行單親二倍體檢測。
本研究患者由于涉及的片段比較大,幾乎涉及到整條10號染色體短臂,且sSMC占比達到100%,依照ACMG和Clingen制定的CNV致病性判定指南,判定該CNV為致病性,該sSMC即為患兒致病原因。本例患兒是由于母親的染色體易位,形成sSMC,在卵子形成過程中沒有參與細胞分裂,遺傳至患兒,核型分析顯示該sSMC是由10號染色體的短臂和13號染色體短臂隨體組成。本病例這種新的sSMC(10)細胞遺傳學形成方式在以往尚未見報道。該患兒未見明顯的結構畸形,進行了腦部核磁檢測,留下了珍貴的病例資料。患兒腦部發現Dandy-Walker畸形等,這在以往尚未見報道,于以后的臨床遺傳學工作是一種啟示。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
海峽科技與產業(2016年3期)2016-05-17 04:32:12
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
