肺原發性黏膜相關淋巴組織結外邊緣區淋巴瘤5例臨床病理分析
2022-03-29 10:31:18孫雪騏黃麒睿
臨床與實驗病理學雜志 2022年1期
孫雪騏,黃麒睿,彭 芳
肺原發性黏膜相關淋巴組織結外邊緣區(primary pulmonary extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue, MALT)淋巴瘤是原發于肺內淋巴組織的惡性淋巴瘤,占肺原發淋巴瘤的70%~90%,但占肺部原發腫瘤的比例不足0.5%[1],臨床較少見,一般為體檢發現,易誤診。本文收集5例肺原發性MALT淋巴瘤,回顧性分析其臨床病理學特征、診斷及鑒別診斷,并介紹預后及分子免疫治療方法,旨在提高對其的認識水平。
1 材料與方法
1.1 材料收集贛州市人民醫院病理科2019年1月~2020年10月活檢確診的5例肺原發性MALT淋巴瘤,5例均經兩位資深病理醫師復核確診。收集患者的臨床資料并電話隨訪患者預后。
1.2 方法標本均經10%中性福爾馬林固定,常規石蠟包埋,3 μm厚切片,HE染色。免疫組化染色采用EnVision兩步法,行高溫抗原修復,DAB顯色等步驟。一抗包括CD20、CD3、CD21、Ki-67、CD10、BCL-6、MUM1、BCL-2、CD5、CD23、Cyclin D1、CK、PAX-5、C-myc,均購自福州邁新公司,操作步驟嚴格按照試劑盒說明書進行,并設陽性和陰性對照。
2 結果
2.1 臨床特征5例患者中男性3例,女性2例,男女比為1 ∶1.5,年齡42~59歲,中位年齡47歲。發生部位:右肺4例(上葉2例,下葉1例,上、下葉合并病灶1例),左肺下葉1例。4例為體檢時發現肺部包塊,1例為入院期間檢查時偶然發現。CT示片狀團塊或斑片狀高密度影,其中3例臨床誤診為原發性肺癌,1例考慮為炎性病變,1例真菌感染。5例臨床均行胸腔鏡下肺葉或肺結節切除術,患者術后均進行隨訪,5例均存活,恢復良好,目前均無復發及淋巴結累及(表1)。
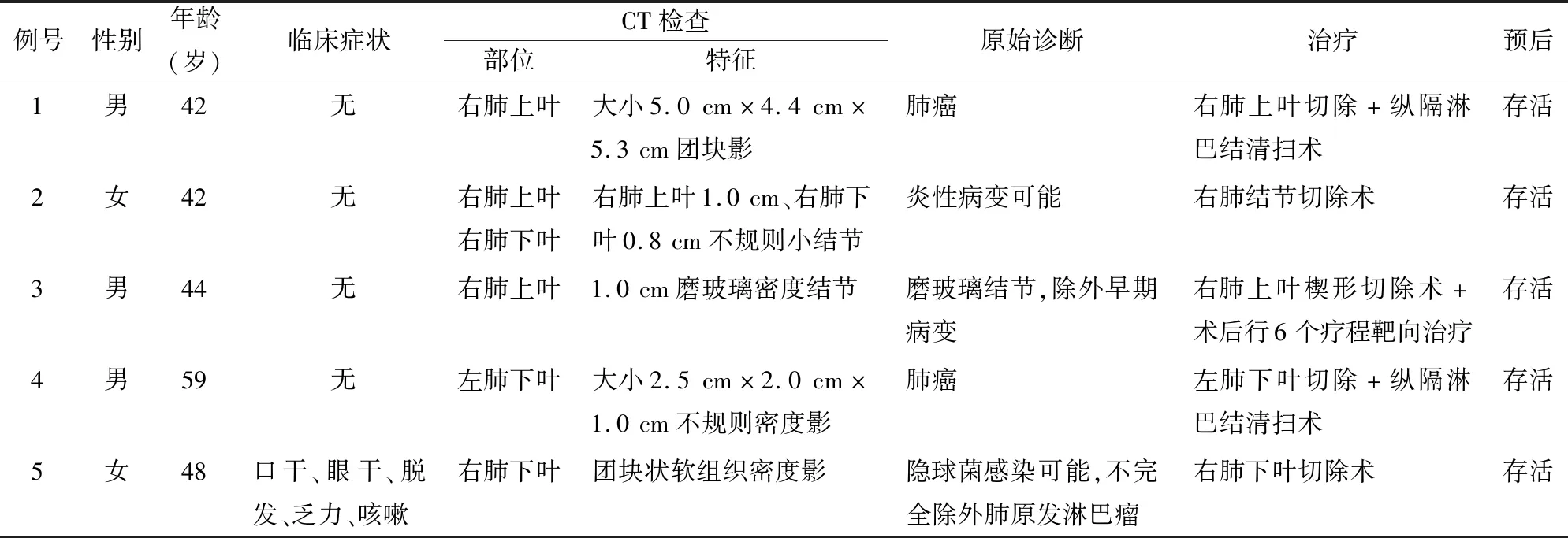
表1 5例肺原發性MALT淋巴瘤患者的臨床資料
2.2 病理檢查
2.2.1眼觀 5例均為大小不等的腫塊,與周圍組織邊界欠清,切面灰白、灰褐色,腫物大小1.2 cm×1.0 cm×1.0 cm~6.0 cm×5.0 cm×5.0 cm。
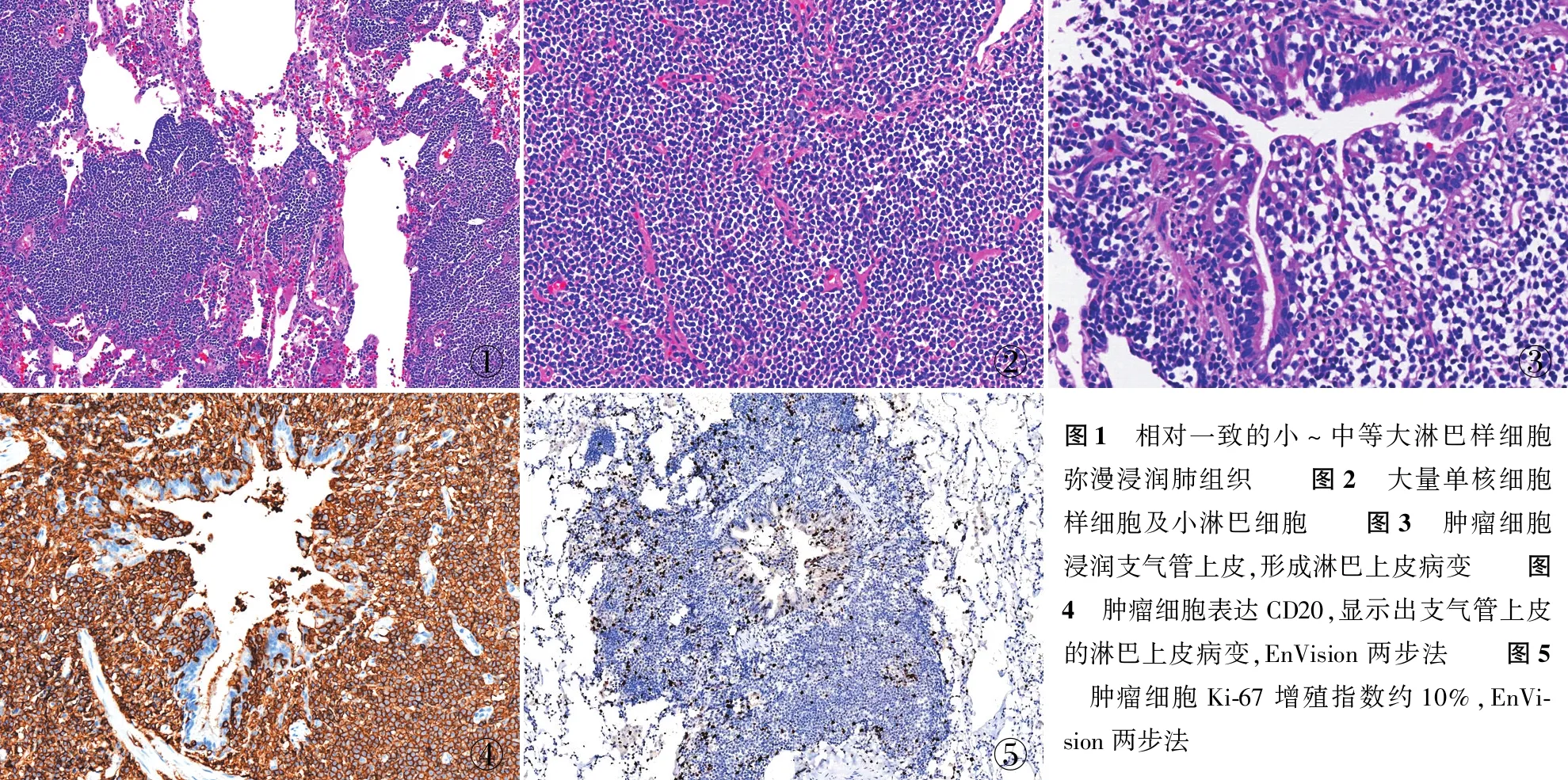
2.2.2鏡檢 5例鏡下形態基本一致,低倍鏡下均以小至中等大小的小淋巴樣細胞彌漫性浸潤為特征(圖1),高倍鏡下瘤細胞形如單核細胞樣細胞和小淋巴細胞(圖2),其間散在分布少許免疫母細胞及中心母細胞樣細胞,部分病例可見漿細胞分化。當3個或以上腫瘤細胞浸潤支氣管、細支氣管黏膜和肺泡上皮,可形成淋巴上皮病變(圖3)。
2.3 免疫表型5例CD20(圖4)、CD79a、PAX-5、BCL-2均陽性,CD3、CD5、Cyclin D1、CD10、BCL-6和CD30均陰性。CD20染色可突出淋巴上皮病變,Ki-67增殖指數為3%~20%(圖5)。統一本組Kappa/Lambda比率來評判單克隆增生的診斷(Kappa ∶Lambda>2.5~3或Lambda ∶Kappa≥1),其中3例顯示輕鏈限制性表達,另2例Kappa、Lambda顯示陽性細胞比例相當。
3 討論
MALT淋巴瘤是一種結外淋巴瘤,占所有B細胞淋巴瘤的7%~8%[2]。正常成人肺部淋巴組織稀少(除自身免疫性疾病如干燥綜合征外),肺部原發性MALT淋巴瘤相對較少見,僅占肺原發性腫瘤的0.5%[1]。MALT淋巴瘤是淋巴組織在抗原作用下克隆性增生所致,肺MALT淋巴瘤被認為是MALT淋巴瘤繼發于炎癥或自身免疫過程中出現的,病因目前尚未明確[3]。盡管目前MALT淋巴瘤分子遺傳學改變尚未得到全基因組測序的證實,但已有研究發現,API2-MALT1基因融合是MALT淋巴瘤中最經典的分子改變[4]。
3.1 臨床特征MALT淋巴瘤好發于成年人,中位年齡61歲,女性稍多于男性。最常見的臨床表現為無癥狀的患者影像學檢查發現腫塊,有癥狀的患者表現為咳嗽、呼吸困難、胸痛和咯血[5]。當腫瘤發生遠處擴散時,優先擴散到其他黏膜部位而非淋巴結[6]。本組5例患者中位年齡(47歲)小于文獻報道的年齡,可能與體檢發現較早相關,且均為影像學檢查發現,與文獻報道一致。
3.2 病理特征MALT淋巴瘤的肉眼典型特征是白色腫塊,邊界不清,質地柔軟,缺乏特異性。鏡下MALT淋巴瘤的組織學特征是淋巴細胞浸潤,腫瘤性B細胞主要存在于反應性濾泡的邊緣區,并延伸到濾泡間區域,這些淋巴濾泡由細胞學形態多樣的小細胞組成,包括小圓形淋巴細胞、中心細胞樣細胞或單核細胞樣細胞。腫瘤細胞侵犯細支氣管或肺泡上皮,可觀察到上皮內腫瘤細胞的植入,導致淋巴上皮病變[7]。
3.3 免疫表型MALT淋巴瘤的腫瘤細胞是單克隆B細胞,免疫組化標記CD20、CD79a和BCL-2陽性,腫瘤B細胞CD10、CD23和BCL-6均陰性,Ki-67增殖指數通常較低(<20%),生發中心內可見大量Ki-67陽性細胞。CD43在某些情況下陽性,但不具有特異性。極少數病例CD5可能陽性[8]。Kappa或Lambda可呈輕鏈限制性表達[9],B細胞或漿細胞中的輕鏈限制性表達高度提示克隆性腫瘤增生[10]。本組重復免疫組化結果顯示,3例顯示輕鏈限制性表達,另2例Kappa、Lambda顯示陽性細胞比例相當。在某些情況下,免疫組化可能由于血清輕鏈、與濾泡樹突狀細胞的反應性或非特異性染色而表現出高背景,而對其中1條或2條輕鏈的敏感性可能較低[11]。漿細胞有豐富的胞質輕鏈,但無表面輕鏈,在識別克隆性B細胞群體方面,流式細胞術比免疫組化、原位雜交和IgH基因重排檢測更敏感[12]。有研究側重于免疫組化和原位雜交檢測對Kappa和Lambda輕鏈的效用,這些研究僅僅是得出正確診斷的一個因素,反應過程則很少被報道有輕鏈限制,因此存在輕鏈限制性表達可能不能明確診斷淋巴瘤,必須結合患者的臨床和其他組織病理學特征進行診斷[13-14]。
3.4 鑒別診斷由于MALT淋巴瘤發病率較低,且組織形態學特征不明顯,特別是穿刺活檢的小標本,易誤診或漏診,因此MALT淋巴瘤通常都是排他性診斷,主要需與以下幾種腫瘤性與非腫瘤性疾病鑒別。(1)其他小B細胞淋巴瘤:如濾泡性淋巴瘤、套細胞淋巴瘤、小淋巴細胞性淋巴瘤和淋巴漿細胞性淋巴瘤,MALT淋巴瘤細胞成分多樣,免疫組化標記Cyclin D1、CD10、BCL-6表達缺失有助于排除套細胞淋巴瘤和濾泡性淋巴瘤,CD5和CD23表達缺失可排除慢性淋巴細胞白血病/小淋巴細胞淋巴瘤[1, 15]。近來有研究報道免疫組化新指標MNDA和IRTA1對鑒別MALT淋巴瘤和其他類型的B細胞淋巴瘤具有診斷意義[16]。MNDA在鑒別MALT淋巴瘤和濾泡性淋巴瘤中有很高的特異性[17]。IRTA1對MALT淋巴瘤具有高度特異性,但目前尚無合適的IRTA1抗體可供商業使用,因此限制了IRTA1免疫組化試劑未被廣泛應用于常規臨床應用。(2)彌漫性淋巴組織增生和淋巴細胞性間質性肺炎:淋巴細胞性間質性肺炎為間質性肺炎的一種,病變與周圍組織分界不清,呈移行關系,淋巴細胞較雜,呈多克隆性增生并伴有不同程度的濾泡間纖維化,PCR檢測顯示免疫球蛋白基因無克隆性重排,在反應條件下也無輕鏈限制性表達[18]。(3)慢性非特異性炎癥反應[7]:其與MALT淋巴瘤均可見小淋巴細胞浸潤,MALT淋巴瘤常呈膨脹性浸潤,與周圍組織分界清楚,且MALT淋巴瘤的小B細胞浸潤超出反應性淋巴濾泡增生的程度,具有沿支氣管血管和小葉間隔擴散的特征性淋巴管模式,淋巴浸潤使肺泡間隔變寬[19]。
3.5 預后及治療MALT淋巴瘤患者預后良好,5年總存活率超過80%,中位生存期超過10年[20]。肺原發性MALT淋巴瘤進展的中位時間為5~6年,肺外受累和淋巴結受累是不良預后因素[6]。MALT淋巴瘤的治療包括手術、化療、免疫治療和放療[21]。研究[22-26]提示免疫調節藥物(the immunomodulatory drugs, IMiDs)合并利妥昔單抗(抗CD20單克隆抗體)對MALT淋巴瘤患者的治療有效果。
綜上所述,肺原發性MALT淋巴瘤較少見,為低度惡性腫瘤,進展緩慢,起病隱匿,無特異性,常為影像學檢查偶然發現,手術完整切除是首選的治療方式。對于無法行手術切除的患者可以選用化療、免疫治療及放療等手段。肺原發MALT淋巴瘤的標準化治療方案仍需深入探討。
