內向整流鉀2.1通道蛋白對人牙囊細胞成骨分化的影響及其機制研究
2022-03-31 09:44:42張鵬左東川牟思宇鐘宇彤袁小平曾錦
華西口腔醫學雜志 2022年2期
張鵬左東川牟思宇鐘宇彤袁小平曾錦
1.西南醫科大學附屬口腔醫院正畸科,瀘州646000;
2.西南醫科大學口頜面修復重建和再生實驗室,瀘州646000;
3.西南醫科大學電生理學教育部重點實驗室,心血管醫學研究所,瀘州646000
基于干細胞的組織工程為組織的修復與再生提供了全新的思路[1-2],但目前調節干細胞生物學特性的眾多分子生物學機制尚不清楚,嚴重阻礙了組織工程的研究進展[3]。牙囊細胞是一種來源于牙囊組織的間充質干細胞,可分化形成牙周膜、牙骨質和牙槽骨[4]。鑒于牙囊細胞在牙周組織分化發育中的特點、顯著的多向分化潛能以及在臨床上較易獲得,使其成為了一種值得考慮的種子細胞[5]。
鉀離子是機體內重要的電解質之一,是維持可興奮細胞靜息膜電位超極化的重要的物質基礎,對細胞增殖、遷移、分化、新陳代謝、滲透壓等生理功能具有廣泛的調控作用。內向整流鉀(inward rectifier potassium,Kir)2.1通道蛋白屬于Kir家族,由KCNJ2基因編碼,是維持可興奮性細胞節律性、傳導性以及興奮性的主要鉀離子通道。生理條件下細胞內鉀離子濃度約為140 mmol·L-1,而細胞外鉀離子濃度約為4.5 mmol·L-1。可興奮細胞如心肌細胞等表達Kir2.1通道,鉀離子順著化學濃度差,經Kir2.1通道介導,由細胞內流向細胞外,從而維持細胞靜息膜電位超極化。Kir2.1通道調控細胞生理學功能的基礎在于其具有典型的內向整流特性,其表現為從鉀平衡電位超極化的內向電導增大,而去極化時外向電導減小,產生一個特征性的負斜率電導區,這一特性決定了其對細胞靜息膜電位超極化的維持[6]。Kir2.1的內向整流特性對鉀離子濃度具有高度依賴性,當細胞外鉀離子濃度升高時,電流-電壓曲線會沿電壓軸向右平移,產生跨交現象,外向電流不減小反而增大,導致細胞膜電位去極化[7]。已有研究表明,Kir2.1通道在非興奮性細胞也發揮了重要的作用。例如,Zhang等[8]報道,抑制Kir2.1通道的功能會引起晚期內皮祖細胞膜電位去極化,從而促進細胞分化;Komarova等[9]以及Weidema等[10]報道Kir
2.1通道通過維持細胞膜電位的穩定參與破骨細胞生理功能的調控。目前在間充質干細胞上已發現包括鈣激活鉀通道、電壓門控鉀通道在內的多種鉀通道,均有功能性的表達[11-12],但目前Kir2.1通道在間充質干細胞的生理功能尚不明確。本研究以人牙囊細胞(human dental follicle cell,hDFC)為研究對象,探討Kir2.1通道對間充質干細胞成骨分化的影響及其機制,以期為應用hDFC修復重建牙周組織提供理論基礎。
1 材料和方法
1.1 主要試劑
α-MEM低(高)糖培養基、磷酸緩沖鹽溶液(phosphate buffered saline,PBS)、胎牛血清(fetal bovine serum,FBS)(Gibco公司,美國),胰蛋白酶(上海碧云天生物技術有限公司),Ⅰ型膠原酶(合肥白鯊生物科技有限公司),青霉素-鏈霉素溶液、4%多聚甲醛固定液、成骨誘導液(Cyagen公司,美國),氯化銫(cesium chloride,CsCl)、4-甲氧基芐基-1-萘基甲基-胺鹽(ML133)(Abmole公司,美國),電解質(Sigma公司,美國),堿性磷酸酶顯色和活性測定試劑盒、Trizol試劑、二甲基亞砜(dimethyl sulfoxide,DMSO)(上海碧云天生物技術有限公司),瓊脂糖(北京亞米生物科技有限公司),總RNA提取試劑盒(北京天根生化科技有限公司),一步法逆轉錄聚合酶鏈反應(reverse transcription-polymerase chain reaction,RT-PCR)擴增試劑盒、定量逆轉錄聚合酶鏈反應(quantitative reverse transcription PCR,RT-qPCR)試劑盒(Toyobo公司,日本)。成骨誘導液的組成成分為10%FBS、10 mmol·L-1β-甘油磷酸鈉、10 nmol·L-1地塞米松、50μg·mL-1抗壞血酸、10 nmol·L-1維生素D3。
1.2 實驗方法
1.2.1 細胞的分離、培養及鑒定 西南醫科大學附屬口腔醫院倫理委員會批準(倫理審批號:2021-1118005)hDFC培養,所有研究對象知情同意。選取因正畸需要,于西南醫科大學附屬口腔醫院口腔頜面外科門診部拔除的牙根發育未完全的第三磨牙牙胚中的牙囊組織,于無菌條件下,立刻收集于含有10%青霉素-鏈霉素雙抗的α-MEM培養基中,在超凈工作臺中,用含有20%雙抗的PBS反復沖洗干凈,換干凈無菌剪剪碎成1 mm3的組織塊,收集于離心管中離心,去上清,加入1 mLⅠ型膠原酶于37℃恒溫水浴中消化30 min,消化期間每隔10 min適當震蕩促使充分消化,消化完成后可見組織塊呈絮狀,再加入等量的含有10%FBS的α-MEM培養液終止消化,離心,去上清,吸取組織塊均勻鋪于培養瓶底部,倒置,加入含有15%FBS的α-MEM培養液3 mL,30 min后翻瓶,于37℃、5%CO2的恒溫孵育箱中培養,大約培養5~7 d可見細胞從組織塊周圍爬出,通過連續傳代培養,細胞被分離擴展用于后續實驗。流式細胞術檢測hDFC的免疫表型,主要檢測hDFC與間充質干細胞相關表面標記物,包括CD73、CD90、CD105、CD31、CD45、CD3(上海艾博抗貿易有限公司),流式細胞術采用Beckman Coulter Cytomics FC 500 MPL系統(Beckman Coulter公司,美國)。
1.2.2 Kir2.1通道蛋白在hDFC中的表達情況 1)RT-PCR檢測Kir2.1的編碼基因KCNJ2在hDFC的表達:hDFC傳至第三代時,均勻接種于小皿中(密度為每孔2×104個細胞),培養細胞7 d后,使用Trizol法提取總RNA(試驗中嚴格執行無酶操作),提取完成后,取1μL樣品使用分光光度計檢測RNA的濃度和純度。采用一步法RT-PCR擴增試劑盒,用提取的RNA為模板,在冰浴中加入RT-PCR反應混合物,反應體系如下:緩沖液5μL,基因正向引物(10μmol·L-1)2μL,基因反向引 物(10μmol·L-1)2μL,逆 轉 錄 酶0.5μL,RNA酶抑制劑0.5μL,熱啟動DNA聚合酶0.5μL,模板RNA 2μL,去RNA酶水37.5μL,反應體系總共為50μL,在PCR儀上按下列條件進行逆轉錄反應,條件設定:45℃30 min、94℃5 min、(94℃30 s、60℃30 s、72℃1 min循環40次)、72℃10 min,反應結束后取5~10μL進行瓊脂糖凝膠電泳檢測。
2)RT-qPCR檢測Kir2.1在hDFC成骨誘導前后KCNJ2的基因表達量:hDFC傳至第三代時,均勻接種于小皿中(密度為每孔2×104個細胞),待細胞生長到60%~70%時,分別對對照組(含10%FBS的α-MEM低糖培養基)和成骨誘導分化(osteogenic differentiation,OS)組(加入1.5 mL成骨誘導液的α-MEM低糖培養基)細胞進行培養,每3 d換液1次,每次1.5 mL,第七天時,提取總RNA和測量其濃度及純度,取10μL變性,變性后的RNA通過一步法RT-PCR擴增試劑盒逆轉錄成互補脫氧核糖核酸(complementary DNA,cDNA),逆轉錄完成后加入等體積的ddH2O稀釋,通過RT-qPCR試劑盒進行擴增。以甘油醛-3-磷酸脫氫酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)為內參,根據公式F=2-ΔΔCT計算目的基因(KCNJ2)的相對表達量,ΔΔCT=(試驗組目的基因平均CT值-試驗組內參基因平均CT值)-(對照組目的基因平均CT值-對照組內參基因平均CT值),其中目的基因KCNJ2的引物序列為:F5’-TGCCCGATTGCTGTTTTC-3’和R5’-GGCTGTCTTCGTCTATTT-3’。
1.2.3 膜片鉗技術檢測hDFC在成骨誘導分化前、后的細胞膜電位變化 hDFC傳至第三代時,在培養基中加入成骨誘導液對hDFC進行成骨誘導分化,于誘導前、誘導7 d后,采用HEKA膜片鉗系統,在電流鉗模式下連續記錄細胞的膜電位。采用電阻為2~3 mΩ的玻璃電極,形成封接,破膜后進行記錄。記錄hDFC膜電位的電極內液各電解質濃度為140 mmol·L-1氯化鉀、1 mmol·L-1氯化鎂、10 mmol·L-1乙二醇雙(2-氨基乙基醚)四乙酸、1 mmol·L-1三磷酸腺苷二鉀鹽、5 mmol·L-1羥乙基哌嗪乙磺酸(pH=7.4),電極外液各電解質濃度為135 mmol·L-1氯化鈉、5.4 mmol·L-1氯化鉀、2 mmol·L-1氯化鈣、1 mmol·L-1氯化鎂、15 mmol·L-1葡萄糖、10 mmol·L-1羥乙基哌嗪乙磺酸(pH=7.4)。所記錄電生理數據采用IgorPro軟件分析。
1.2.4 膜電位去極化或抑制Kir2.1通道功能對hDFC成骨分化的影響 實驗分為:OS組(成骨誘導液),K+組(成骨誘導液+50 mmol·L-1鉀),CsCl組(成骨誘導液+10 mmol·L-1CsCl),ML133組(成骨誘導液+20μmol·L-1ML133)。
1)茜素紅染色及礦化結節定量分析:hDFC傳至第三代時,均勻種板于6孔板中(密度為每孔2×104個細胞)培養,待細胞生長到60%~70%時,去除基礎培養基,PBS輕柔清洗2~3次,各組細胞于37℃、5%CO2的孵育箱中避光培養,每3 d換液1次,每次1.5 mL,培養14 d后,吸取誘導液,PBS反復沖洗2~3次,吸干,4%多聚甲醛固定細胞15 min,再次用PBS反復輕柔沖洗2~3次,吸干,進行茜素紅染色3 min,PBS沖洗多余染液,于搖床上輕微搖蕩10 min,更換為新PBS,于肉眼下觀察染色情況,并于顯微鏡下采圖,觀察鈣結節形成情況,再行茜素紅染色半定量分析,向各組孔板中加入10%氯化十六烷基吡啶2 mL,充分混勻后,吸取100μL置于96孔板中,于562 nm處下檢測吸光度值。
2)堿性磷酸酶染色及活性測定:hDFC以每孔2×104個細胞的密度接種于6孔板中,待細胞生長到60%~70%時,分別對各組細胞進行成骨分化誘導,每3 d換液1次,每次1.5 mL,第七天時按照試劑盒說明書進行堿性磷酸酶染色及活性測定,用酶標儀在405 nm處測定吸光度,計算堿性磷酸酶活性比。堿性磷酸酶活性的定量數據根據成骨分化第七天的對照結果進行校準。
3)成骨分化相關基因[RUNX相關轉錄因子(Runt related transcription factor,RUNX)2、骨鈣素(osteocalcin,OC)]的表達:hDFC傳至第三代時,均勻接種于小皿中(密度為每孔2×104個細胞),待細胞生長到60%~70%時,分別對各組細胞進行成骨分化誘導,每3 d換液1次,14 d時同樣以上述1.2.2中的方法檢測和計算目的基因RUNX2、OCN的相對表達量,基因引物序列如下:RUNX2:F5’-GCAGCAGCAGCAGCAGGAG-3’和R5’-GCACCGAGCACAGGAAGTTGG-3’,OCN:F5’-GCCAGGCAGGTGCGAAGC-3’和R5’-GTCAGCCAACTCGTCACAGTCC-3’。
1.2.5 鈣離子成像檢測膜電位變化對細胞內鈣離子濃度的影響 hDFC傳至第三代時,細胞均勻接種于小皿中的玻片上(密度為每孔2×104個細胞),于孵育箱中培養1 d,待hDFC貼壁于玻片上,用鈣離子熒光染料(Fura-2/AM)負載細胞30 min,然后用PBS沖洗掉剩余染料,將染色好的細胞玻片置于倒置顯微鏡載物臺檢測皿中,組裝好循環系統,通過循環系統連續灌流標準(含5.4 mmol·L-1鉀)的細胞外液,然后用40倍油鏡觀察并選取hDFC,待hDFC細胞內鈣信號基線穩定后,記錄胞內鈣離子熒光強度,再灌流含有低鉀(2 mmol·L-1)的細胞外液,觀察和記錄鈣信號的改變情況,其中鈣信號采用TILL vision軟件觀察和記錄2種波長340 nm和380 nm激發下熒光強度的比值(F340/380),轉換頻率為0.2 Hz,發射光波長為510 nm,曝光時間為5 ms。
1.3 統計學分析
數據以均數±標準差表示,利用SPSS 17.0軟件進行統計學分析,多組數據統計學分析采用單因素方差分析,P<0.05時記為差異有統計學意義。
2 結果
2.1 hDFC的分離、培養及鑒定
hDFC經原代培養5 d后,觀察可見已具有典型的間充質干細胞形態,hDFC呈長梭形,以放射狀或旋渦狀從組織塊向周圍生長(圖1A)。利用酶消化法進行分離純化后觀察可見,傳至第三代的hDFC形態均一,多呈長梭形,呈魚群狀或放射狀排列(圖1B)。hDFC具有較強的成骨分化能力,成骨誘導14 d后經茜素紅染色,鏡下觀察可見大量紅色礦物結節(圖1C)。流式細胞結果顯示:hDFC表面抗原CD73(97.93%)、CD90(99.96%)和CD105(82.98%)表達量高,而CD31(1.58%)、CD45(3.55%)和CD3(4.36%)表達量極低(圖2),證實培養獲得的hDFC為間充質來源的干細胞。

圖1 hDFC培養及鑒定 ×40Fig 1 Cultivation and identification of hDFCs ×40
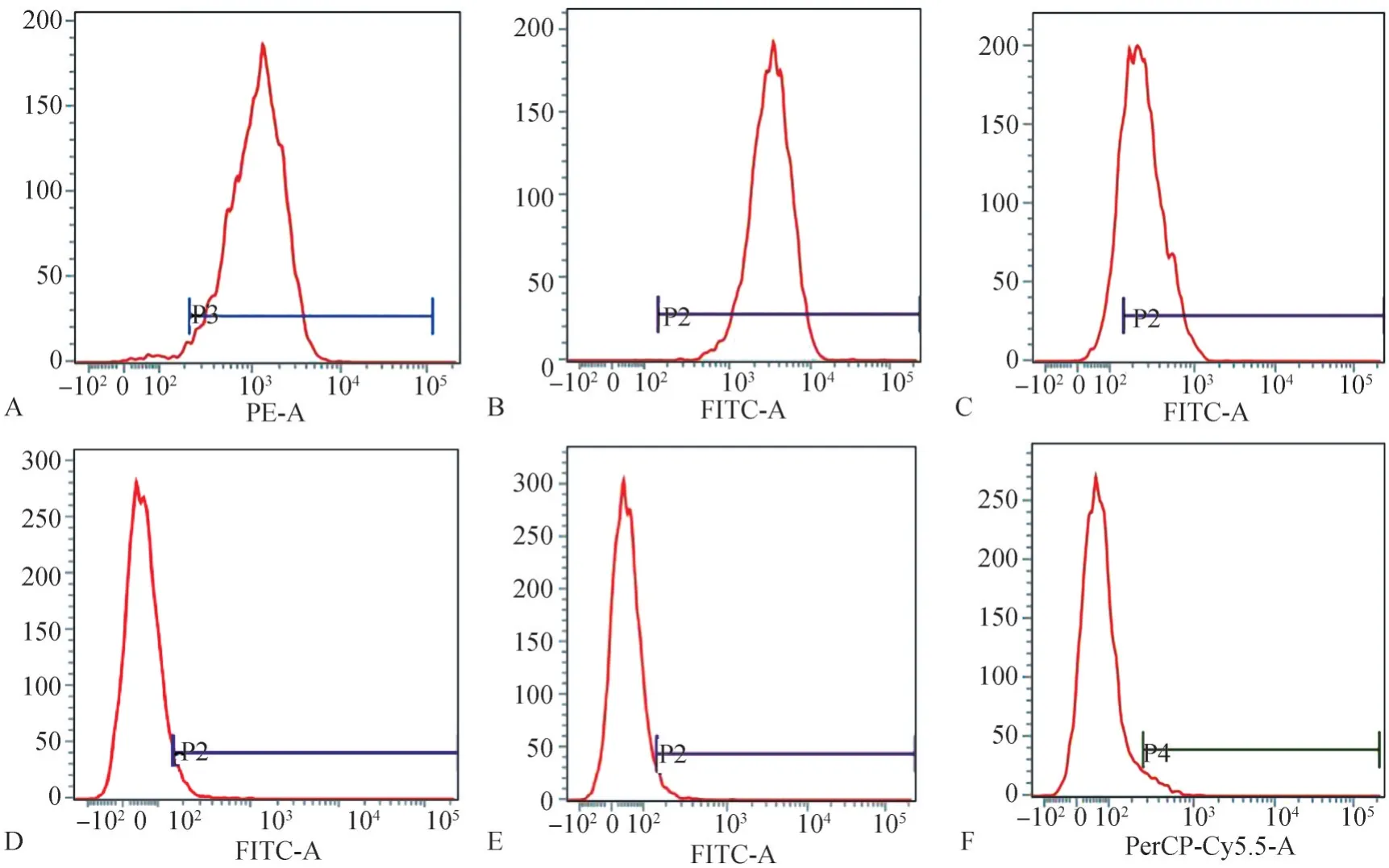
圖2 細胞表面抗原表達的流式細胞術檢測結果Fig 2 Results of cell surface antigen expression by flow cytometry
2.2 Kir2.1通道蛋白在hDFC中的表達情況
RT-PCR結果顯示:hDFC表達Kir2.1基因KCNJ2(圖3A);RT-qPCR結果顯示:與對照組細胞相比較,OS組細胞的Kir2.1基因KCNJ2的表達上調(圖3B)。
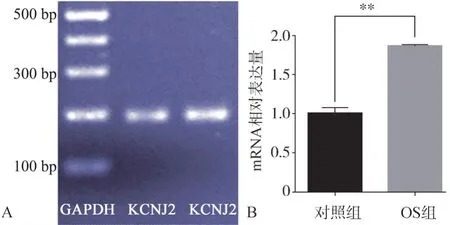
圖3 Kir2.1基因KCNJ2的表達情況Fig 3 KCNJ2 expression of Kir2.1 channel
2.3 成骨誘導前后hDFC的膜電位變化
全細胞膜片鉗結果顯示,成骨誘導前hDFC的膜電位約為(-12±3.2)mV,成骨誘導后的膜電位約為(-47±5.2)mV(圖4),這一結果說明,hDFC成骨分化的過程中伴隨著細胞膜電位超極化的發生。
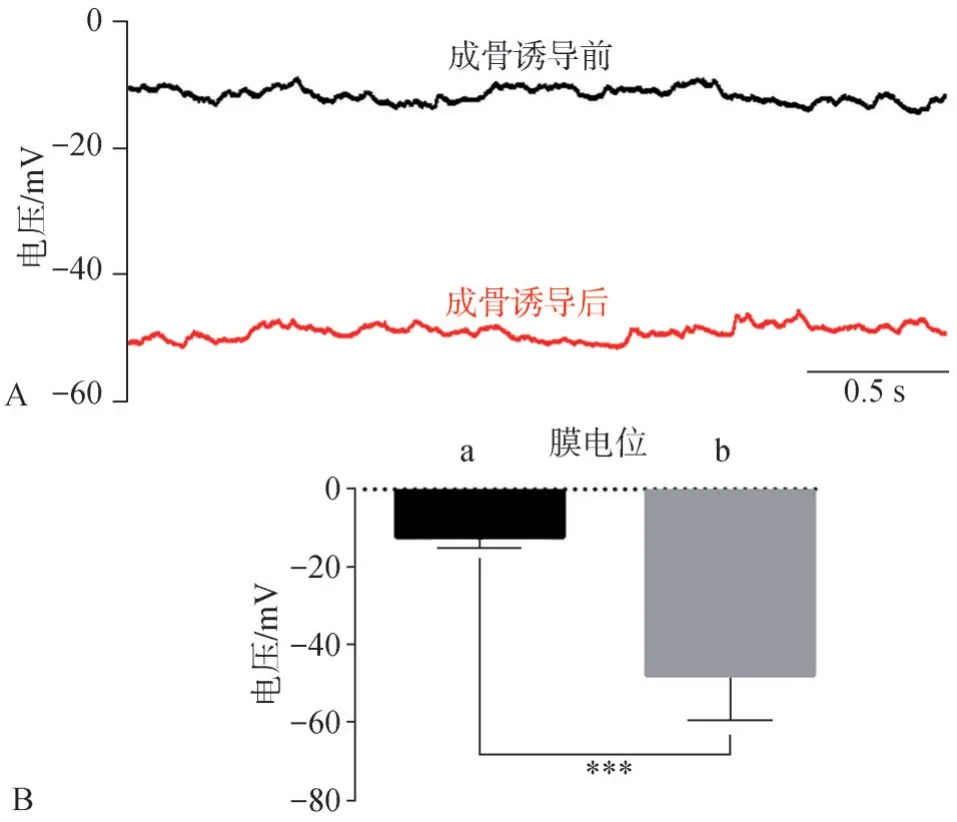
圖4 hDFC成骨誘導前后的膜電位變化Fig 4 Membrane potential changes of the hDFCs before and after osteogenic induction
2.4 膜電位去極化或抑制Kir2.1通道功能對hDFC成骨分化的影響
茜素紅染色及礦化結節定量分析結果顯示:經成骨誘導14 d后,OS組細胞鏡下觀察可見大量紅色礦物結節,而K+組、CsCl組和ML133組細胞的紅色礦物結節數量明顯減少。肉眼觀察結果見圖5A~D;鏡下觀察結果見圖5E~H,茜素紅染色半定量分析見圖6。
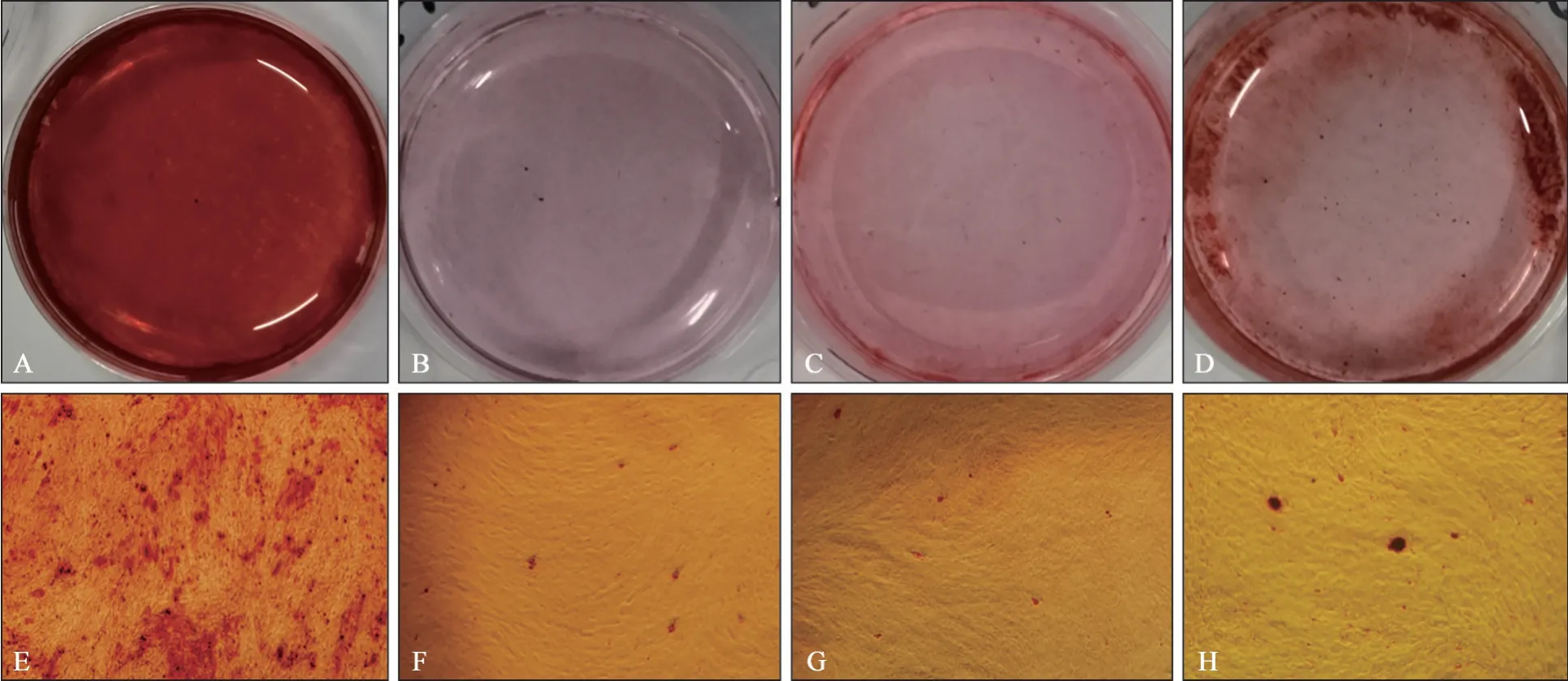
圖5 茜素紅染色結果 ×40Fig 5 Resultsof alizarin red staining ×40
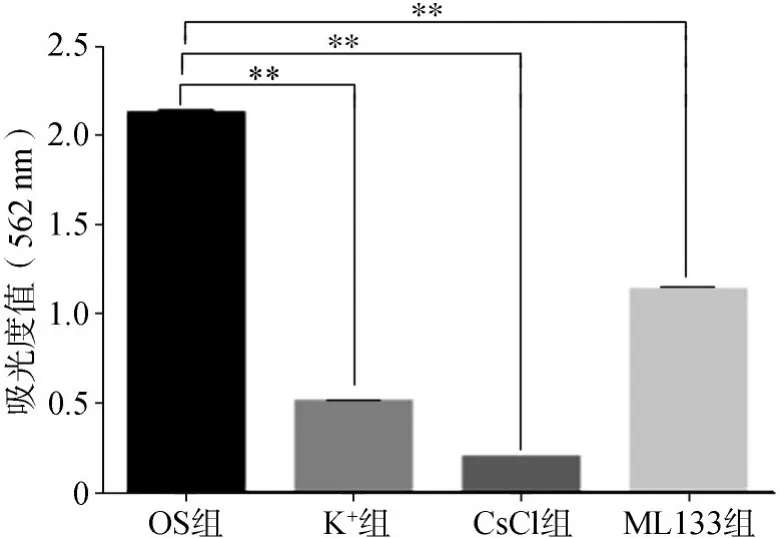
圖6 茜素紅染色半定量分析結果Fig 6 Semi-quantitative analysis result of alizarin red staining
堿性磷酸酶染色結果顯示:經成骨誘導7 d后,與OS組細胞相比較,K+組CsCl組和ML133組細胞的堿性磷酸酶活性明顯降低,肉眼觀察結果見圖7A~D,鏡下觀察結果見圖7E~H,堿性磷酸酶活性測定分析結果見圖8。

圖7 堿性磷酸酶染色結果 ×40Fig 7 Resultsof alkalinephosphatasestaining ×40
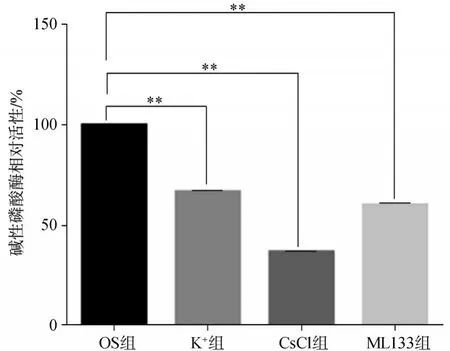
圖8 堿性磷酸酶活性測定結果Fig 8 Activity measurement results of alkalinephosphatase
2.5 成骨分化相關基因的表達情況
RT-qPCR結果顯示:經成骨誘導14 d后,與OS組細胞相比較,K+組、CsCl組、ML133組細胞的RUNX2、OCN基因表達明顯降低(圖9)。
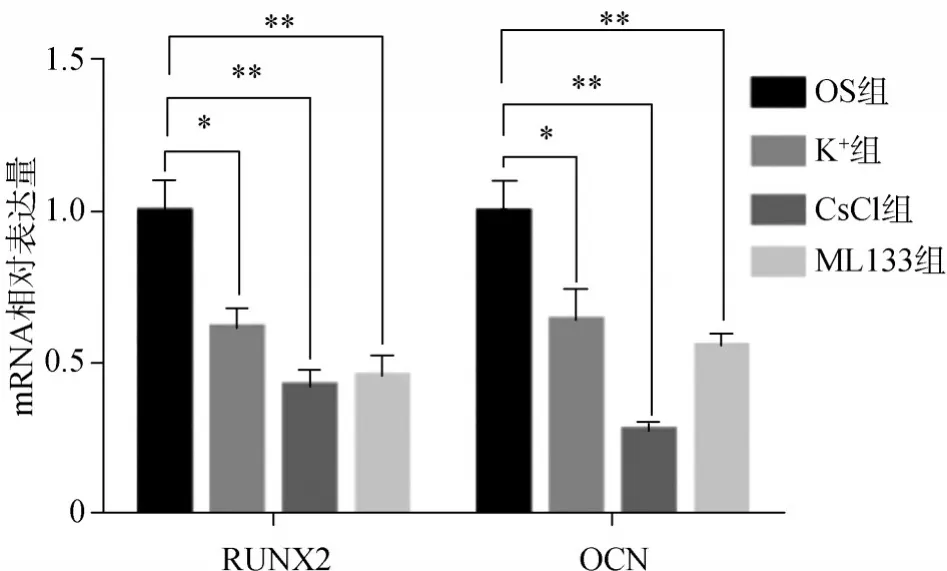
圖9 RT-qPCR檢測成骨分化相關基因RUNX2、OCN的表達情況Fig 9 Detection of osteogenic differentiation-related genes RUNX2 and OCN expression by RT-qPCR
2.6 膜電位超極化對hDFC胞內鈣離子濃度的影響
鈣離子成像結果顯示,當細胞外鉀離子的濃度從5.4 mmol·L-1降為2 mmol·L-1時,hDFC胞內鈣離子熒光強度顯著增強(圖10),F340/380的比值見圖11,上述結果表明,膜電位超極化會引起hDFC胞內鈣離子濃度的升高。
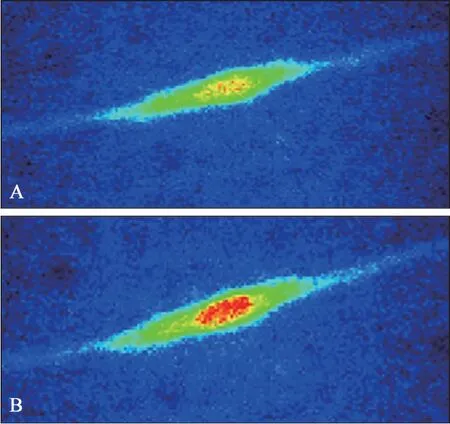
圖10 鈣離子熒光染色結果 ×40Fig 10 Results of calcium ion fluorescent staining ×40
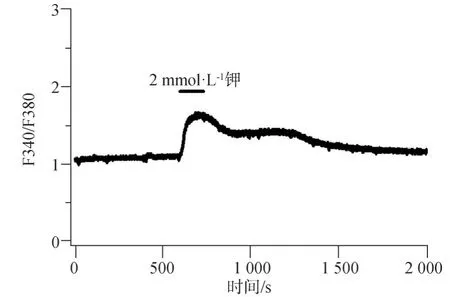
圖11 F340/380比值的分析結果Fig 11 The analysis result of F340/380 ratio
3 討論
自2005年Morsczeck等[13]從人的牙囊組織中首次分離出hDFC以來,該細胞因其較好的干細胞特性以及再生牙周組織的潛力而受到廣泛關注。
本研究中的hDFC提取于因正畸需要而拔除的健康第三磨牙牙胚,經原代培養后具有典型的間充質干細胞形態,通過流式細胞術鑒定細胞表面標志物,證明其為純化的間充質干細胞。hDFC是來源于牙囊組織的間充質干細胞,具有較強的成骨分化潛能,本研究培養獲得的hDFC在成骨誘導后,經茜素紅染色可見大量的成骨礦化結節,這與以往相關的報道[14]一致。
有研究表明,間充質干細胞的成骨分化過程受細胞膜電位的調控。例如,Kirkham等[15]報道,通過電刺激的方法使人骨髓間充質干細胞膜電位超極化能夠促進細胞成骨分化;Sundelacruz等[16]和Bhavsar等[17]報道,人骨髓間充質干細胞在成骨分化的過程中伴隨著膜電位的超極化,并且他們發現,通過提高細胞外鉀離子的濃度使細胞膜電位去極化,能夠抑制細胞的成骨分化過程。本研究中,首先檢測了hDFC成骨分化過程中膜電位的變化;膜片鉗結果顯示,細胞成骨誘導前的膜電位約為(-12±3.2)mV,經成骨誘導7 d后,膜電位約為(-47±5.2)mV,這一結果說明,hDFC成骨分化的過程中伴隨著膜電位的超極化;其次,觀察了膜電位去極化對hDFC成骨分化的影響,提高細胞外鉀離子的濃度是使細胞膜電位去極化常用的方法[16]。
本研究結果顯示,OS組細胞經成骨誘導后可見大量礦化結節,而提高細胞外鉀離子的濃度后,細胞的成骨礦化能力顯著降低。上述結果說明,細胞膜電位超極化的發生在hDFC成骨分化的過程中起到了重要的作用。
Kir2.1通道蛋白在興奮性細胞發揮了至關重要的作用,但近年來越來越多的證據顯示,Kir2.1通道蛋白在非興奮性細胞如內皮細胞、神經膠質細胞、多能干細胞等也發揮了重要的作用。如Kito等[18]報道,Kir2.1通道蛋白的表達上調在內質網應激引起的腦毛細血管內皮細胞死亡過程中起到了重要的作用;Gattlen等[19]報道,抑制Kir2.1通道蛋白在小膠質細胞中的表達,能夠抑制細胞的增殖能力從而減輕神經痛。
Pini等[20]發現,Kir2.1通道功能的喪失影響多能干細胞成骨和軟骨形成過程。在本研究中,通過RT-PCR的方法首先證明了hDFC表達Kir2.1,并且成骨誘導后Kir2.1的基因表達量明顯上調;其次,應用Kir2.1通道選擇性阻斷劑(CsCl)和特異性阻斷劑(ML133)都證明了抑制Kir2.1通道的功能能夠抑制hDFC的成骨礦化能力。因此,根據以往文獻報道以及本研究的實驗結果,證明了Kir2.1通道蛋白介導的膜電位超極化在hDFC成骨分化的過程中起到了關鍵的作用。Kirkham等[15]和Pchelintseva等[21]在人間充質干細胞的研究表明,膜電位超極化可以引起細胞內鈣離子濃度的升高繼而調控成骨分化,本研究中確實發現膜電位超極化能夠引起hDFC細胞內鈣離子濃度的升高,這一結果與以往在間充質干細胞的研究相一致。
牙槽骨缺損的修復、再生一直是牙周組織工程研究的熱點,牙囊細胞作為牙周組織工程的種子細胞,具有間充質干細胞的特征,表現出強大的多向分化能力及再生能力[22],是骨缺損修復值得考慮的種子細胞。有學者[23]報道Kir2.1通道蛋白的功能與成骨密切相關,基因突變導致的Kir2.1功能喪失,臨床上會出現安德森綜合征,臨床表現之一為骨骼發育異常[20]。本研究的研究結果也表明抑制Kir2.1通道蛋白的功能可以抑制hDFC成骨分化。在將來的動物實驗中,可針對Kir2.1通道蛋白這一靶點進行調控,從而為促進骨缺損的修復、再生提供理論基礎。
利益沖突聲明:作者聲明本文無利益沖突。
