TSC2基因突變致兒童肺淋巴管平滑肌瘤病1例并文獻復習
2022-05-17 11:23:22王世遠謝新星秦海燕
現代醫藥衛生 2022年9期
關鍵詞:基因突變
王世遠,彭 昌,謝新星,秦海燕
(遵義醫科大學附屬醫院兒內科,貴州 遵義 563000)
肺淋巴管平滑肌瘤病(PLAM)于1937年首次提出,并于1986年在中國首次報道。該病是一種病因未明,由平滑肌異常增殖導致支氣管、淋巴管和小血管阻塞,呈進行性發展的全身性疾病[1]。PLAM患者肺部最易受累,常表現為彌漫性間質性肺疾病。該病好發于育齡婦女,關于兒童PLAM的相關報道較少見。本文報道了1例TSC2基因突變致兒童PLAM。
1 臨床資料
患兒,男,6歲。因“咳嗽2周,加重2 d”就診于本院。就診前2周無明顯誘因出現咳嗽,呈陣發性串咳,無刺激性嗆咳,無氣促、喘息及呼吸困難,無發熱、胸痛及胸悶,無嘔吐、腹瀉,無腹脹及腹痛,無頭痛、頭暈及抽搐。首次發病就診于當地縣醫院,診斷為支氣管肺炎,予以頭孢噻肟抗感染及止咳化痰等對癥支持治療,4 d后咳嗽好轉出院。入本院前2 d,再次出現咳嗽加重,安靜狀態下無明顯氣促及呼吸困難,無喘息及發紺,無胸悶、胸痛及心悸,無發熱、嘔吐及頭痛。體格檢查:體溫36.7 ℃,脈搏96次/分,呼吸25次/分,血壓98/48(1 mm Hg=0.133 kPa),體重17 kg;神志清楚,精神好,顏面口唇無發紺,全身皮膚無瘀點、瘀斑及出血點,全身未捫及腫大淋巴結;雙肺呼吸音粗、對稱,可聞及少許細濕啰音及哮鳴音;心率96次/分,心律齊,心界無擴大,各瓣膜聽診區未聞及病理性雜音;腹部及神經系統未見異常,雙下肢無水腫。患兒既往體健,無相關疾病史。患兒父母均健康,無家族遺傳病史。實驗室檢查:血常規、尿常規及糞便常規、電解質、肝腎功能、心肌酶、C反應蛋白、凝血功能、D二聚體、免疫球蛋白(Ig)G、IgA、IgM、IgE、補體C3、C4均未見異常。風濕十二項檢查示:抗SM抗體陽性,抗核抗體(1∶100)弱陽性(核顆粒型),余未見異常。細胞免疫功能檢查示:總T淋巴細胞絕對值672 μL-1(正常參考值:955~2 860 μL-1),T輔助細胞絕對值375 μL-1(正常參考值:550~1 440 μL-1),T抑制細胞絕對值219 μL-1(正常參考值:320~1 250 μL-1),其余未見異常。支原體抗體1∶320陽性,氧分壓70.4 mm Hg,二氧化碳分壓26.8 mm Hg,pH 7.363,實際碳酸氫根14.9 mmol/L,標準碳酸氫根16.9 mmol/L,堿剩余9.3 mmol/L,提示輕度低氧血癥,代謝性酸中毒合并呼吸性堿中毒(代償期)。纖維支氣管鏡檢測示氣管支氣管內膜炎,肺泡灌洗液涂片見多量中性粒細胞及小吞噬細胞,肺含鐵血紅素細胞檢測陽性,肺泡灌洗液培養未見異常,未見腫瘤細胞。肺活檢示急慢性炎癥細胞浸潤。真菌(1-3)-B-D-葡聚糖、乙型肝炎抗體、人類免疫缺陷病毒抗體、丙型肝炎抗體、梅毒抗體均為陰性。頭顱CT、骨髓細胞學檢查及培養未見異常。胸部高分辨CT示:雙肺廣泛性囊性病變。見圖1。肺功能示混合性通氣功能障礙。
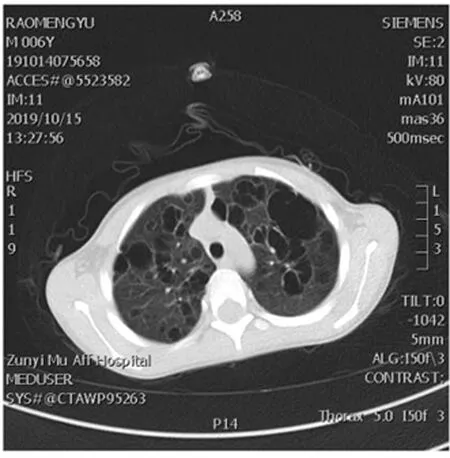
圖1 患兒胸部高分辨率CT圖(2019年9月15日)
經患兒父母知情同意后分別抽取患兒及其父母靜脈血2 mL送檢。采用第一代測序技術進行全外顯子測序(由北京康旭醫學檢驗所完成),本實驗共進行12次聚合酶鏈反應(PCR)擴增及24個基因序列測定反應。檢測結果示:患兒及其母親的TSC2基因10號外顯子c.856A>G,p.Met286Val雜合突變;患兒父親TSC2基因未見突變。采用突變致病性預測軟件SIFT、Polyphen 2、MutationTaster分析發現,TSC2基因10號外顯子c.856A>G,p.Met286Val突變。Polyphen 2及MutationTaster軟件預測結果顯示:該位點突變的致病性為可能致病,SIFT軟件預測結果顯示為中性。見表1和圖2。

表1 PCR擴增突變位點
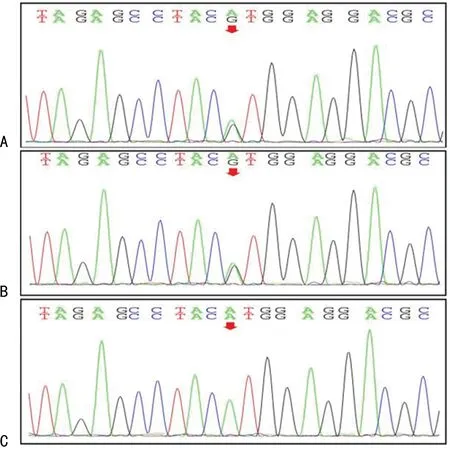
A.患兒,雜合突變;B.患兒母親,雜合突變;C.患兒父親,未見突變。
患兒經常規抗炎、止咳、祛痰等對癥支持治療后咳嗽好轉出院,其家屬拒絕進一步治療。3個月后患兒再次因咳嗽氣促2 d繼發肺部感染入本院,復查胸部CT示:雙肺囊性病變加重,并發雙肺氣胸。見圖3。經對癥處理后患兒病情好轉出院。目前仍在隨訪當中。
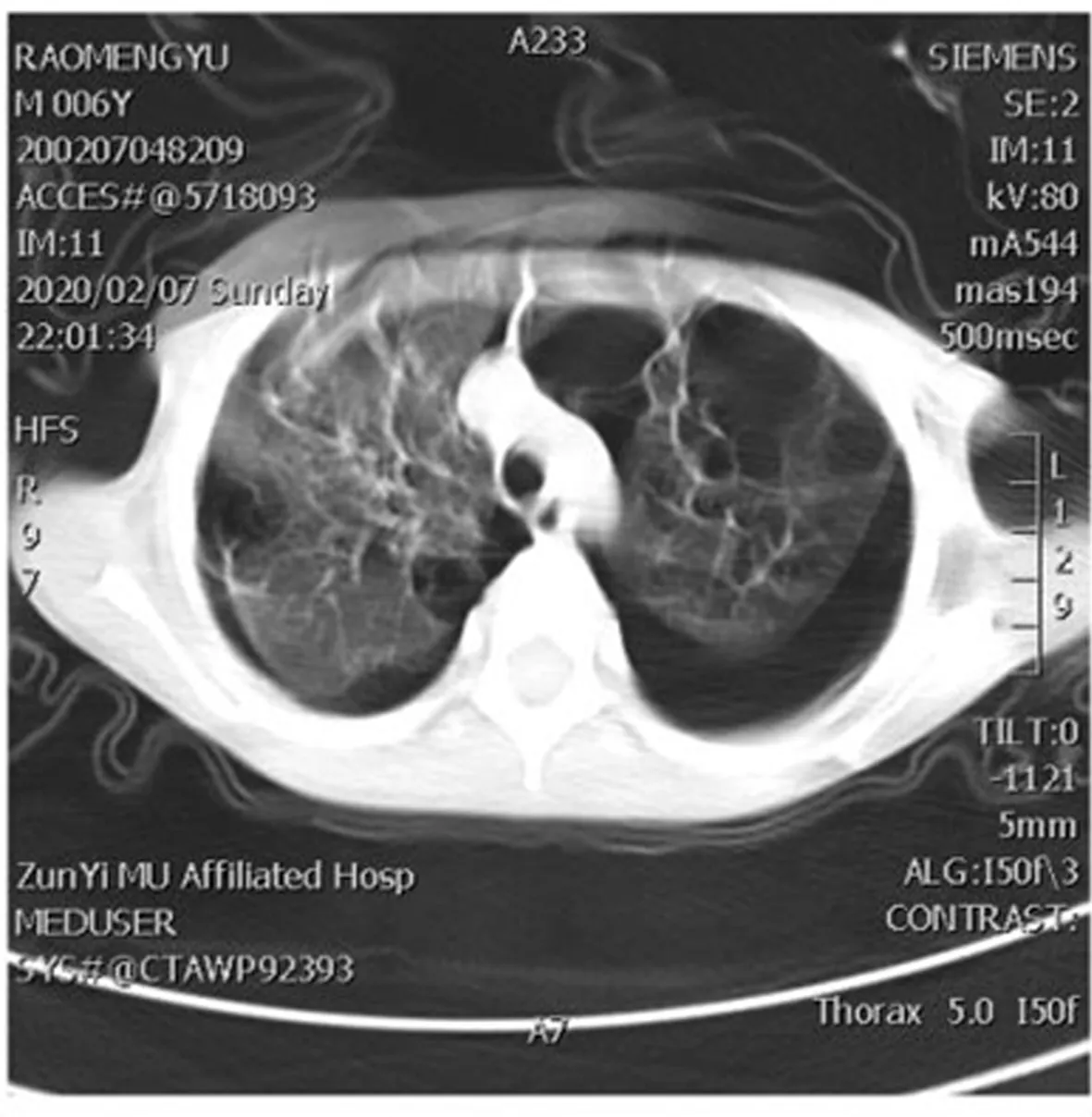
圖3 患兒高分辨率CT圖(2020年2月7日)
2 討 論
PLAM是一種累及多系統的罕見病,好發于育齡婦女。流行病學調查發現,無遺傳疾病婦女發生PLAM的概率約為百萬分之2.6,其中1/3的患者合并結節性硬化[2]。PLAM早期臨床癥狀無特異性,主要表現為進行性呼吸困難、咳嗽、氣胸、咯血、乳糜胸等,晚期可發生呼吸衰竭。
目前,PLAM發病機制尚不清楚。研究表明,PLAM的病因與TSC1和TSC2基因密切相關[3]。TSC1定位于人類9號染色體長臂3區4帶,編碼錯構瘤蛋白;TSC2定位在16號染色體短臂13區3帶,編碼馬鈴薯球蛋白。當TSC1或TSC2基因發生突變時,雷帕霉素(mTOR)信號通路靶點激活,導致肺內平滑肌樣細胞過度生長、增殖[4]。TSC2基因突變頻率高于TSC1基因,且TSC2基因突變患者病情也通常比TSC1基因突變患者更嚴重。TSC2基因9號外顯子中的c.856A>G具有致病性[5]。本例患兒為TSC2基因10號外顯子c.856A>G,p.Met286Val雜合突變,該突變遺傳自母親。c.856A>G,p.Met286Val的致病性評估在不同預測工具中的結果不一致,但該突變位點的致病性在成人患者中有相關報道[5]。因此推斷該位點突變存在致病性。本例患兒的母親并沒有發病,表明該基因的突變只是PLAM發生的一個條件,可能還有其他某些未知因素參與其中。
TSC1/TSC2屬于腫瘤抑制基因,二者編碼蛋白形成復合體,通過多個信號通路參與調節細胞增殖、生長。TSC基因有大量突變類型,主要包括錯義、無義、剪切、移碼及大片段缺失突變5種類型[6]。TSC2基因體細胞突變參與散發型淋巴管肌瘤病的發生,而結節性硬化癥分型與TSC1、TSC2基因突變有關[7]。有研究表明,PLAM與TSC2基因雜合體缺失及突變密切相關[8]。目前已明確淋巴管平滑肌瘤病(LAM)與TSC2基因的突變有關,該基因的蛋白質產物tuberin是mTOR復合物(mTORC)活性的介體,tuberin蛋白功能的喪失會導致mTORC1的激活失控[9]。同時,mTORC1的激活導致真核起始因子4E結合蛋白1、S6激酶和核糖體S6蛋白磷酸化,從而促進蛋白質翻譯、細胞生長和增殖[10]。本研究發現TSC2基因10號外顯子出現雜合突變,但該位點突變導致PLAM的具體機制不清楚。
PLAM主要病理改變為肺泡囊性病變,肺淋巴管平滑肌細胞增生會引起血管、淋巴管和細支氣管管壁狹窄甚至閉塞,導致氣胸或乳糜胸。病變組織內可見一定程度的含鐵血黃素,這是由于擴張和扭曲的小靜脈破裂引起少量出血而造成的。因此,本例患兒肺泡灌洗液中肺含鐵血黃素染色陽性,不考慮肺含鐵血黃素沉著癥。本例患兒肺活檢僅見到肺組織中炎癥細胞浸潤,未提示典型的PLAM病理特征,其原因可能是患兒年齡小,或取材位置不典型所致。胸部高分辨CT對PLAM的診斷有較高價值。本研究采用胸部高分辨CT檢查可見肺內彌漫性多發圓形或類圓形薄壁囊腔病灶,病灶邊界清楚,囊腔病灶大小為0.2~4.0 cm,所有囊腔病灶的囊腔壁厚度小于2 mm,囊壁光滑且厚度較均勻。2016年美國胸科學會/日本呼吸學會發布的《LAM診斷和管理臨床指南》提出,血管內皮生長因子-D水平大于800 pg/mL且具有典型LAM的肺部高分辨CT特征患者可診斷為PLAM,但檢出率只有70%[11]。因此,PLAM確診仍依賴肺活檢。目前,PLAM的治療暫無有效方法,早期主要為對癥治療。研究表明,mTOR信號通路靶點激活是導致PLAM的一個重要因素,而西羅莫司可抑制mTOR的活化,阻斷LAM細胞增殖,從而達到治療PLAM的目的。晚期肺移植可能是一種有效方法。
綜上所述,兒童PLAM可通過臨床特點、影像學檢查及相關基因檢測協助診斷。本研究發現的1例兒童PLAM是由TSC2基因10號外顯子c.856A>G,p.Met286Val雜合突變引起,這一發現豐富了基因突變譜。PLAM是一種多系統的慢性疾病,常出現在疾病的晚期。早期診斷和早期干預可大大提高患者生活質量,延長患者生存時間。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
