Peutz-Jeghers綜合征雙胞胎患兒病例報道并文獻復習
2022-05-17 11:23:22于敬紅于淑鳳宋振鳳陳云慶王彩霞
現代醫藥衛生 2022年9期
于敬紅,于淑鳳,宋振鳳,趙 燕,陳云慶,王彩霞△
(青島大學附屬醫院:1.兒童醫學中心;2.病理科,山東 青島 266555)
Peutz-Jeghers(P-J)綜合征是一種常染色體顯性遺傳病,由STK11(LKB1)基因突變引起,主要臨床表現為皮膚黏膜色素沉著和胃腸道多發錯構瘤。本文報道的1例先證者為雙胞胎次子,因便血及腹疼就診,查體可見典型皮膚色素沉著,胃腸鏡檢查發現典型息肉,外周血及口腔黏膜細胞全外顯子基因檢測提示STK11基因突變。先證者胞兄臨床表現及胃腸鏡檢查較先證者輕,STK11基因未見異常,其父母無P-J綜合征相關臨床表現,外周血和口腔黏膜細胞基因檢測未見STK11突變,內鏡檢查亦未見特征性息肉。本例先證者突變基因及基因突變方式較少見,現報道如下。
1 臨床資料
先證者,男,12歲,為雙胞胎次子,因“間斷便血15 d伴腹痛2 d”于2020年10月31日入當地醫院。15 d前先證者無明顯誘因出現大便表面帶血,為暗紅色,伴有肛門腫物脫出,無惡心、嘔吐,無腹脹、腹瀉,無排便困難及疼痛,腫物于當地醫院回納,未再脫出,大便亦未再帶血。2 d前先證者出現陣發性左上腹脹痛,與進食無明顯關系,持續1~2 min后可自行緩解,無胸悶、憋氣,無心慌、氣短,無惡心、嘔吐,無便血。詢問病史得知先證者出生后口唇即有少許黑色扁平斑點,不凸出于口唇黏膜表面。隨年齡增長斑點漸增多,部分融合成片,且手指末端出現黑斑,顏色逐漸加深,未予診治。入院后查體:先證者神志清,精神好,口唇黑斑,部分融合成片,未突出皮面,口腔黏膜可見1處黑斑,手指散在黑斑(圖1),腹部柔軟,無包塊,左下腹壓痛,心肺查體未見明顯異常。
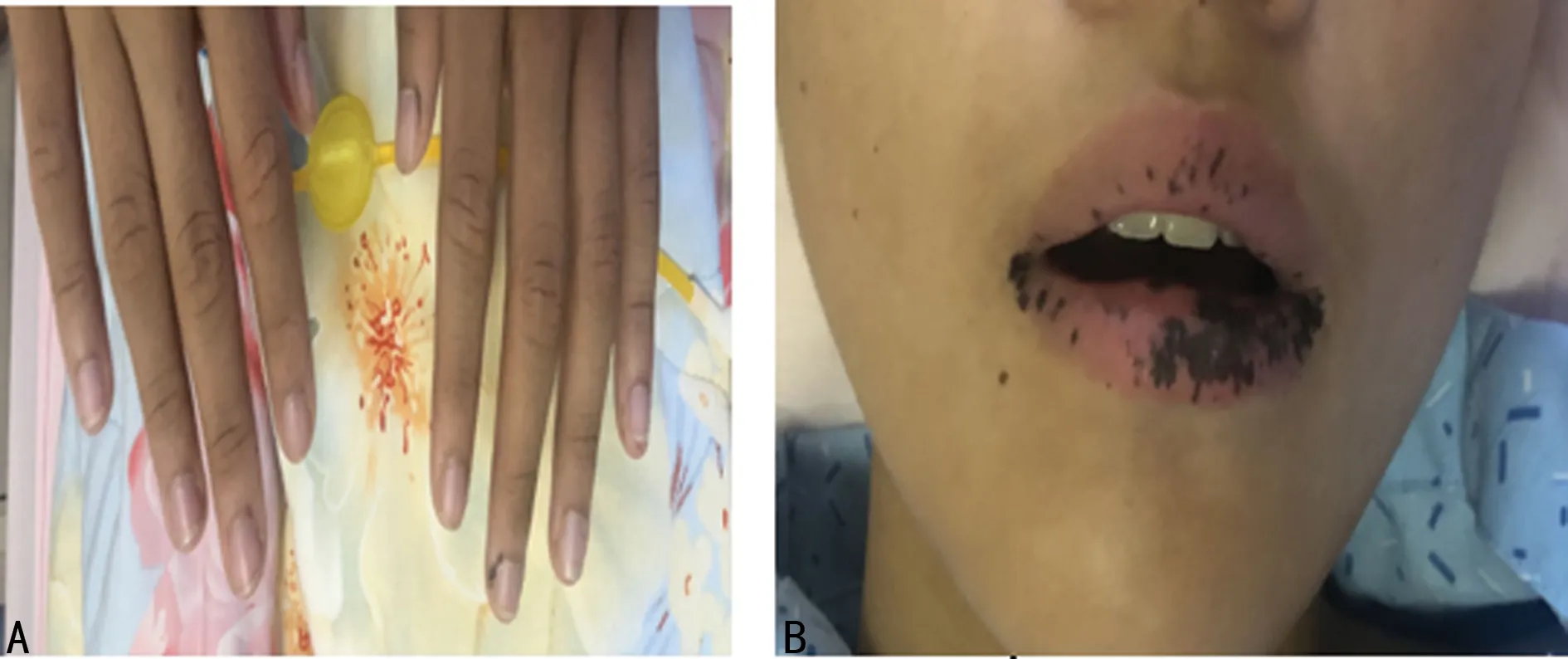
A.手指黑斑;B.口唇黑斑。
先證者入院完善相關檢查,排除禁忌證后行結腸鏡及胃鏡檢查。內鏡下可見胃及腸內多發息肉,直徑2~20 mm,形狀以半球型為主,部分呈結節分葉狀(圖2A、2B)。病理提示:胃增生性息肉并管狀腺瘤,十二指腸降段增生性息肉,腸黏膜組織呈息肉樣增生。為進一步明確小腸內息肉增生情況,行小腸CT造影,結果示小腸多發息肉(圖2C)。治療過程中,先證者出現腸套疊(小腸-小腸型,圖2D),及時予腹腔鏡手術切除套疊腸管,并送病理。免疫組織化學檢查示:CK7陰性,CK20陽性,MUC5AC陰性,MUC2部分陽性,結蛋白陽性,平滑肌肌動蛋白陽性,D2-40陰性,Ki-67部分陽性。在先證者監護人均知情同意并經過醫院倫理委員會(批件號:QYFYWZLL26508)批準下,完善外周血及口腔黏膜細胞全外顯子基因檢測,提示基因STK11發生c.291-1G>A突變(chr19:1218416-1218416),且為低比例嵌合,導致剪接位點發生改變,與常染色體顯性遺傳病P-J綜合征相關。因先證者確診P-J綜合征,而先證者胞兄具有相同的口唇及十指黑斑,為明確診斷入本院。先證者胞兄無明顯不適,查體可見口唇及十指散在黑色斑點,未突出皮面,無融合,較先證者輕。行小腸CT造影示部分性中腸旋轉不良,排除禁忌證后完善胃腸鏡檢查。內鏡下可見散在大小不等息肉,直徑可達10 mm,形狀以半球形為主。胃部病理示腺體性息肉,腸道病理示增生性息肉。對先證者胞兄外周血及口腔黏膜細胞行全外顯子基因檢測均為野生型,未見STK11基因突變。見圖3。
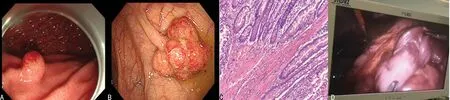
A.胃鏡下部分息肉表現;B.腸鏡下部分息肉表現;C.息肉病理表現(200×);D.先證者腹腔鏡下腸套疊。
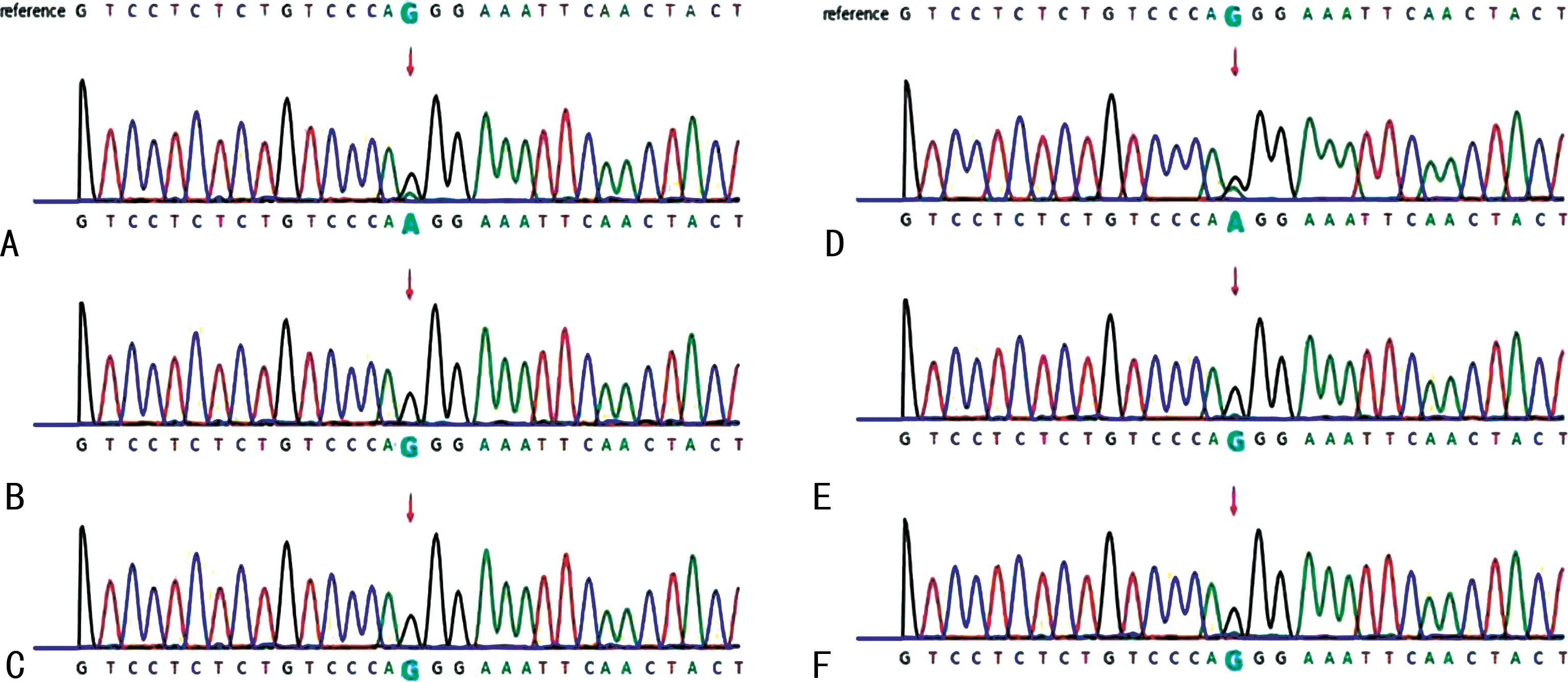
A.先證者外周血基因檢測;B.先證者父親外周血基因檢測;C.先證者母親外周血基因檢測;E.先證者口腔拭子基因檢測;F.先證者胞兄外周血基因檢測;G.先證者胞兄口腔拭子基因檢測。
對先證者3代親屬進行調查,未見與先證者具有相同或相似臨床表現的家屬,未發現因消化道息肉行相關手術或有相關惡性腫瘤家屬。同時對先證者父母進行外周血全外顯子基因檢測,未發現STK11基因突變。先證者胞兄臨床表現輕,內鏡下行胃腸道息肉切除后未見明顯不適,大便顏色、形狀正常。先證者因小腸套疊行腹腔鏡部分腸管切除術,術后恢復好,未見明顯不適。出院后定期隨訪雙胞胎患兒恢復情況,目前雙胞胎患兒病情穩定,無不適。
2 討 論
P-J綜合征以皮膚黏膜色素沉著和胃腸道多發性錯構瘤息肉為主要臨床表現,其發生率為1/8 600~1/25 000[1]。皮膚黏膜色素沉著主要位于口唇、肛周、手指或足趾,1歲以后顏色加深,除口腔黏膜外,其他部位色素沉著均在青春期以后逐漸消失[2]。息肉主要位于胃腸道,近幾年也有鼻、膽囊、輸尿管等消化道以外息肉的報道[3-4]。P-J綜合征患者特征性臨床表現出現時間不一,有的出生后即存在,有的則在成年后出現。國外對P-J綜合征特征性臨床表現出現的中位時間進行研究時發現,該病出現的時間與先證者為家族遺傳還是散發病例無關[5]。兒童時期的主要臨床表現為息肉導致腸套疊、貧血、梗阻等,成人時期則面臨各系統患癌風險的增加[6]。本例先證者即因肛門腫物脫出就診而發現。
P-J綜合征為臨床罕見病,關于指導該病管理的臨床資料和科學數據較少見。隨著基因工程的不斷發展,醫學界對P-J綜合征認識不斷深入,其臨床診斷標準不斷更新。1997年,P-J綜合征臨床診斷標準簡單描述為:(1)胃腸道典型息肉或胃腸道有1個息肉;(2)伴有P-J綜合征典型色素沉著或P-J綜合征家族史。2010年更新為:有2個或以上組織學典型P-J綜合征息肉;(2)或近親中P-J綜合征家族史個體有任何數量的典型息肉;(3)或近親中P-J綜合征家族史個體有特征性黏膜皮膚色素沉著及任何數量的P-J綜合征息肉[7]。當然確切診斷還需行基因檢測,明確有STK11基因突變。P-J綜合征息肉的形成主要與轉化生長因子β(TGF-β)相關。LKB1基因突變導致TGF-β不能正常表達,使其失去調節控制上皮細胞增殖作用,從而形成息肉。關于息肉治療,歐洲消化道內窺鏡協會建議,對于大于8歲的無癥狀P-J綜合征患者,應每1~3年完善1次胃腸道全面檢查,若小腸典型息肉直徑為>15~20 mm時應進行選擇性息肉切除術,以防止腸套疊[4]。國內有關P-J綜合征患者的研究顯示,患者黏膜色素斑極少發生惡變。研究者在追蹤多例P-J綜合征患者疾病轉歸情況時發現,患者胃腸道腫瘤發生率較普通人明顯升高,且乳腺、膀胱、食管等腸外腫瘤發生率也會增加。
STK11基因位于常染色體19p13.3,共含有10個外顯子,其中外顯子1~9為基因編碼區,10為非翻譯區。有研究利用直接測序或多重連接擴增技術發現,P-J綜合征患者常見的基因突變主要發生在外顯子1~9[8]。2018年關于P-J綜合征基因突變種類已報道400余種[9],主要為點突變和缺失突變,低比例嵌合相對少見,發生在內含子的低比例嵌合更為少見。STK11基因編碼絲氨酸/蘇氨酸激酶,參與控制細胞周期停滯、p53介導的細胞凋亡、細胞外因子和TGF-β信號傳導、ras基因誘導的轉化、能量代謝等[10]。因此,P-J綜合征患者癌變概率增加的主要原因為STK11基因突變,其使抑癌基因P53表達降低,由P53介導的細胞增殖和凋亡失去調控,從而導致異常細胞增殖[11]。STK11基因的氨基端為非催化域,羧基末端為調節域,可使絲氨酸及蘇氨酸被磷酸化,從而發揮磷酸化和RNA翻譯后修飾的作用[5,9]。P-J綜合征雙胞胎患兒相對少見,目前相關研究顯示,雙胞胎患兒無論臨床表現還是基因突變位點、類型均相同[12]。本例先證者外周血及口腔黏膜細胞STK11呈內含子c.291-1G>A的低比例嵌合狀態,且雙胞胎患兒基因不同。
低比例嵌合體指個體體內同時存在兩套基因:一種為正常基因,占絕大部分;另一種為突變基因,所占比例相對較少。當突變基因占據一定比例后可在遺傳表達中發揮作用,影響個體臨床表型。目前,國內外關于P-J綜合征低比例嵌合報道不多見,相關報道也只是粗略描述了患者無相關臨床表現,均因具有P-J綜合征臨床表現的親屬確診而確診[13]。本例先證者雖是低比例嵌合,但具有P-J綜合征特異性臨床表現,且先證者胞兄具有相關臨床表現及胃腸道息肉,但STK11基因卻未見突變。相關研究顯示,P-J綜合征存在明顯的遺傳異質性和表型異質性,一些患者只表現出皮膚黏膜色素沉著,而一些患者既有皮膚黏膜色素沉著,又有胃腸道多發息肉,甚至一些患者臨床表現已經達到P-J綜合征診斷標準,但其STK11基因卻未檢測到突變[14-15]。本研究中,先證者胞兄體現出遺傳與表型異質性特點,推測該例患兒基因嵌合比例更低或存在目前檢測手段尚不能檢測出的突變。各種突變致使DNA在轉錄過程中出現錯譯或無義突變等導致轉錄提前終止,最終致使RNA翻譯的蛋白質鏈變短或蛋白質結構發生改變而失去相應功能。隨著基因工程的不斷發展,在P-J綜合征患者中發現,一些內含子突變同樣致病。其原因為距離外顯子兩端較近的內含子部分堿基序列組成剪接位點,當內含子與外顯子同時被轉錄在一條原始RNA中時,原始RNA通過剪接位點剪切掉內含子片段,而外顯子根據剪切位點會重新組合為成熟mRNA。因此,當外顯子兩端的內含子堿基發生變化時可直接導致剪接位點發生變化,進而導致mRNA堿基序列或堿基數量改變,使翻譯出的蛋白質鏈長度或結構發生改變,從而致病。
綜上所述,P-J綜合征是一種臨床罕見病,具有較低發生率,其發病機制及遺傳特點目前尚未闡明。P-J綜合征患者成年后各系統癌癥發生率較高,因此一旦確診應及時切除息肉,并定期進行內鏡檢查,以改善預后。本研究中,具有P-J綜合征典型臨床表現的同卵雙胞胎患兒基因突變存在差異,雙胞胎長子未檢測出基因突變,而雙胞胎次子基因突變方式為STK11基因內含子低比例嵌合,其父母無P-J綜合征臨床表現,也未檢測出相關基因突變。結合本病例資料,作者推測P-J綜合征可能存在其他基因突變或目前檢測手段難以檢測出的更低低比列嵌合等致病原因。因此,建議對于有疑似P-J綜合征臨床表現的患兒,除進行胃腸鏡、組織病理及血液基因檢測外,可增加病理組織的基因測定,從而為P-J綜合征的研究及指導優生優育提供可靠資料。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
