異甘草素納米混懸劑的制備及其體內藥動學研究
2022-06-15 01:38:24劉勇華張留超郭曉娜
中成藥 2022年5期
劉勇華, 張留超, 郭曉娜
(黃河科技學院,河南 鄭州 450005)
異甘草素是從紅血藤、肉豆蔻、甘草等植物根莖中提取得到的異黃酮類化合物,具有抗腫瘤、抗心律失常、抗炎、抗病毒、保肝、抗氧化等多種藥理作用[1-2],但該成分溶解度較差,從而影響其口服吸收[3]。目前,對異甘草素相關制劑的研究主要有脂質體[4]、聚乳酸納米粒[5]、微乳[6]等,但存在制備過程繁瑣、包封率和載藥量低等缺點。
納米混懸劑可有效提高藥物溶出度和生物利用度[7-10],與固體脂質納米粒、聚合物納米粒比較,具有制備工藝簡單、載藥量大等優勢。馬啟珍等[11]采用磷脂作為穩定劑,制備了異甘草素納米混懸劑,但磷脂在水相中穩定性較差[12],也未進行凍干工藝研究,可能會存在穩定性問題;本實驗采用沉淀-高壓均質法制備異甘草素納米混懸劑,并考察其體內藥動學,以期為相關制劑研究提供參考。
1 材料
1.1 儀器 安捷倫1260型高效液相色譜儀(美國安捷倫公司);ATS型高壓均質機(加拿大SEEKER工業公司);AR2140型電子天平[梅特勒-托利多儀器(上海)有限公司];M-sizer 2000型激光粒度分析儀(英國馬爾文儀器有限公司);Thermo Scientific 88882010型渦旋混合器(美國Thermo Scientific公司);LHS-HC-I型恒溫恒濕箱(上海一恒科學儀器有限公司);SJIA-10N-50型冷凍干燥機(寧波市雙嘉儀器有限公司);ND400型氮氣吹掃儀(杭州瑞誠儀器有限公司)。
1.2 試劑與藥物 異甘草素對照品(批號191105,純度98.2%,天津一方科技有限公司);異甘草素原料藥(批號P20191025T,純度98.0%,武漢東康源科技有限公司)。聚乙烯吡咯烷酮K30(批號25000240379,亞什蘭集團公司);泊洛沙姆188(批號514621-3,德國BASF公司);羧甲基纖維素鈉(CMC-Na,批號181005,佛山市日特化工有限公司)。
1.3 動物 清潔級SD大鼠,體質量(300±20)g,購于河南省動物實驗中心,動物生產許可證號SCXK(豫)2016-0001。
2 方法與結果
2.1 異甘草素含量測定
2.1.1 色譜條件 Agilent Plus C18色譜柱(4.6 mm×150 mm,5 μm);流動相0.2%磷酸-甲醇(55∶45);體積流量1.0 mL/min;柱溫35 ℃;檢測波長372 nm。
2.1.2 線性關系考察 精密稱取異甘草素對照品20 mg,溶于10 mL甲醇中,作為貯備液(2.0 mg/mL);配制0.2%磷酸-甲醇(55∶45)混合溶液,超聲處理10 min,作為稀釋液,將貯備液稀釋至50、5、0.5、0.1、0.05 μg/mL,在“2.1.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程為Y=1 201X-3.084 1(r=0.999 9),在0.05~50 μg/mL范圍內線性關系良好。
2.1.3 供試品溶液制備 取1 mL納米混懸劑至100 mL量瓶中,加入50 mL甲醇超聲提取6 min,冷卻至室溫,稀釋液定容,0.22 μm微孔濾膜過濾,取2 mL續濾液至10 mL量瓶中,稀釋液定容,即得。
2.1.4 方法學考察 取同一份納米混懸劑,按“2.1.3”項下方法平行制備6份供試品溶液,在“2.1.1”項色譜條件下進樣測定,測得異甘草素峰面積RSD為1.26%,表明該方法重復性良好。取同一份供試品溶液,室溫下于0、4、8、12、24、48 h在“2.1.1”項色譜條件下進樣測定,測得異甘草素峰面積RSD為0.67%,表明溶液在48 h內穩定性良好。取同一份供試品溶液,在“2.1.1”項色譜條件下進樣測定6次,測得異甘草素峰面積RSD為0.22%,表明儀器精密度良好。取9份供試品溶液,分為3組,每組3份,分別加入2.5、5.0、7.5 mg對照品,按“2.1.3”項下方法制備供試品溶液,在“2.1.1”項色譜條件下進樣測定,測得異甘草素平均加樣回收率分別為100.38%、99.65%、99.80%,RSD分別為1.09%、0.73%、0.84%。
2.2 納米混懸劑制備 采用沉淀-高壓均質法。取0.5 g異甘草素原料藥,加入40 mL無水乙醇,超聲處理3 min溶解,作為有機相;取穩定劑(PVP K30和/或泊洛沙姆188)適量,加入100 mL蒸餾水,超聲處理3 min溶解,作為水相,在轉速800 r/min、溫度50 ℃的磁力攪拌儀上將有機相緩慢滴加到水相中,滴完后繼續攪拌3 h,在設定的均質壓力下循環均質數次,蒸餾水定容至100 mL,即得。
2.3 單因素試驗
2.3.1 穩定劑種類 固定異甘草素用量0.5 g,藥物與穩定劑比例1∶3,均質壓力100 MPa,均質次數8次,考察PVP K30、泊洛沙姆188、兩者混合物(比例1∶2、1∶1、2∶1)對粒徑、PDI的影響,結果見圖1。由此可知,單用PVP K30或泊洛沙姆188時,粒徑均超過230 nm,PDI均在0.2左右,相對較高;兩者聯用時,粒徑、PDI均降低,其比例為1∶1時更明顯。因此,選擇PVP K30-泊洛沙姆188(1∶1)作為穩定劑。
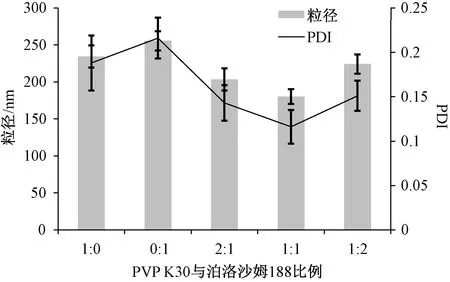
圖1 穩定劑種類對粒徑、PDI的影響(n=3)Fig.1 Effects of stabilizer type on particle size and PDI (n=3)
2.3.2 藥物與穩定劑比例 固定異甘草素用量0.5 g,PVP K30與泊洛沙姆188比例1∶1,均質壓力100 MPa,均質次數12次,考察藥物與穩定劑比例1∶2、1∶2.5、1∶3、1∶3.5、1∶5對粒徑、PDI的影響,結果見圖2。由此可知,隨著藥物與穩定劑比例增加,粒徑呈先降后升的趨勢,可能是由于過多的穩定劑附著在納米粒周圍,導致其粒徑變大;PDI呈先降后趨穩的趨勢,表明穩定劑用量增加有助于提高粒徑分布的集中度,降低PDI。另外,藥物與穩定劑比例為1∶3、1∶3.5時,兩者粒徑、PDI無明顯差異,考慮到載藥量,選擇1∶3作為兩者比例。

圖2 藥物與穩定劑比例對粒徑、PDI的影響(n=3)Fig.2 Effects of drug-stabilizer ratio on particle size and PDI (n=3)
2.3.3 均質壓力 固定異甘草素用量0.5 g,PVP K30與泊洛沙姆188比例1∶1,藥物與穩定劑比例1∶3,均質次數12次,考察均質壓力80、90、100、110、120 MPa對粒徑、PDI的影響,結果見圖3。由此可知,隨著均質壓力增加,粒徑、PDI呈先降后升的趨勢;在110 MPa時粒徑較低,但PDI高于100 MPa時。由于在100、110 MPa下粒徑無明顯差異,故選擇100 MPa作為均質壓力。
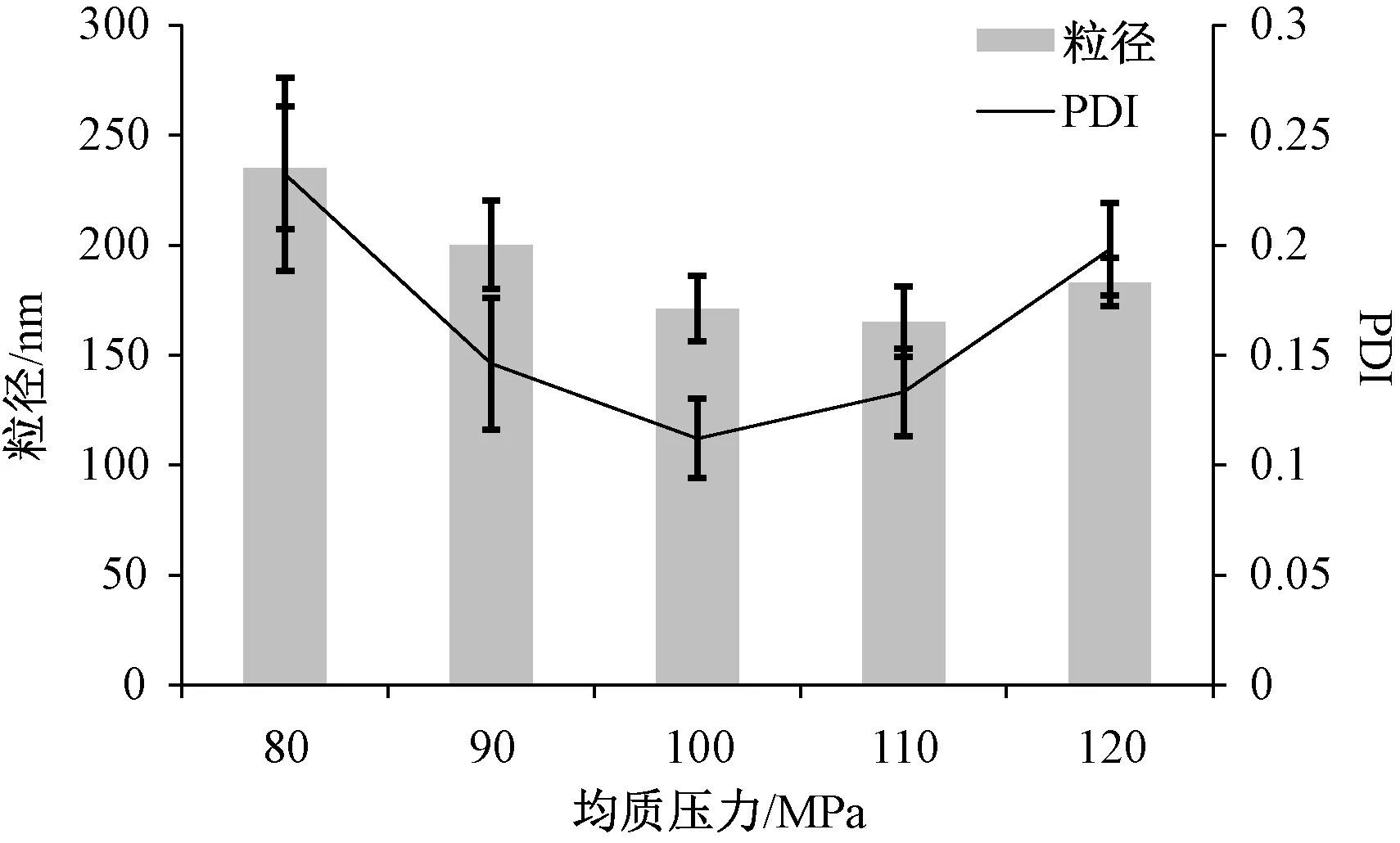
圖3 均質壓力對粒徑、PDI的影響(n=3)Fig.3 Effects of homogenization pressure on particle size and PDI (n=3)
2.3.4 均質次數 固定異甘草素用量0.5 g,PVP K30與泊洛沙姆188比例1∶1,藥物與穩定劑比例1∶3,均質壓力100 MPa,考察均質次數10、11、12、13、15次對粒徑、PDI的影響,結果見圖4。由此可知,均質次數過多時粒徑、PDI反而升高,可能是加劇奧斯瓦爾德熟化作用所致[13]。因此,選擇均質次數為12次,此時粒徑、PDI均較低。

圖4 均質次數對粒徑、PDI的影響(n=3)Fig.4 Effects of homogenization frequency on particle size and PDI (n=3)
2.3.5 工藝確定 取0.5 g異甘草素原料藥,加入40 mL無水乙醇,超聲處理3 min溶解,作為有機相;取穩定劑[PVP K30-泊洛沙姆188(1∶1)]1.5 g,加入100 mL蒸餾水,超聲處理3 min溶解,作為水相,在轉速800 r/min、溫度50 ℃的磁力攪拌儀上將有機相緩慢滴加到水相中,滴完后繼續攪拌3 h,在100 MPa均質壓力下循環均質12次,蒸餾水定容至100 mL,即得。
2.4 載藥量測定 制備3批納米混懸劑,分別精密量取1 mL,在-60 ℃下預凍2 d,置于-25 ℃冷凍干燥機中抽真空,靜置2 d,稱定粉末質量(W1)。取1 mL供試品溶液至100 mL量瓶中,加入50 mL甲醇超聲提取6 min,靜置至室溫,稀釋液定容,0.22 μm微孔濾膜過濾,取2 mL續濾液至10 mL量瓶中,稀釋液定容,在“2.1.1”項色譜條件下進樣測定,計算異甘草素含量(W2),平行3次,計算載藥量,公式為載藥量=(W2/W1)×100%。結果,納米混懸劑平均載藥量為(24.36±0.83)%。
2.5 粒徑、Zeta電位測定 取3份納米混懸劑,每份0.5 mL,加入20 mL蒸餾水稀釋,測定粒徑、Zeta電位,結果見圖5~6。由此可知,平均粒徑、PDI、Zeta電位分別為(163.5±6.8)nm、0.109±0.12、(-34.1±2.6)mV,表明該工藝重復性良好。
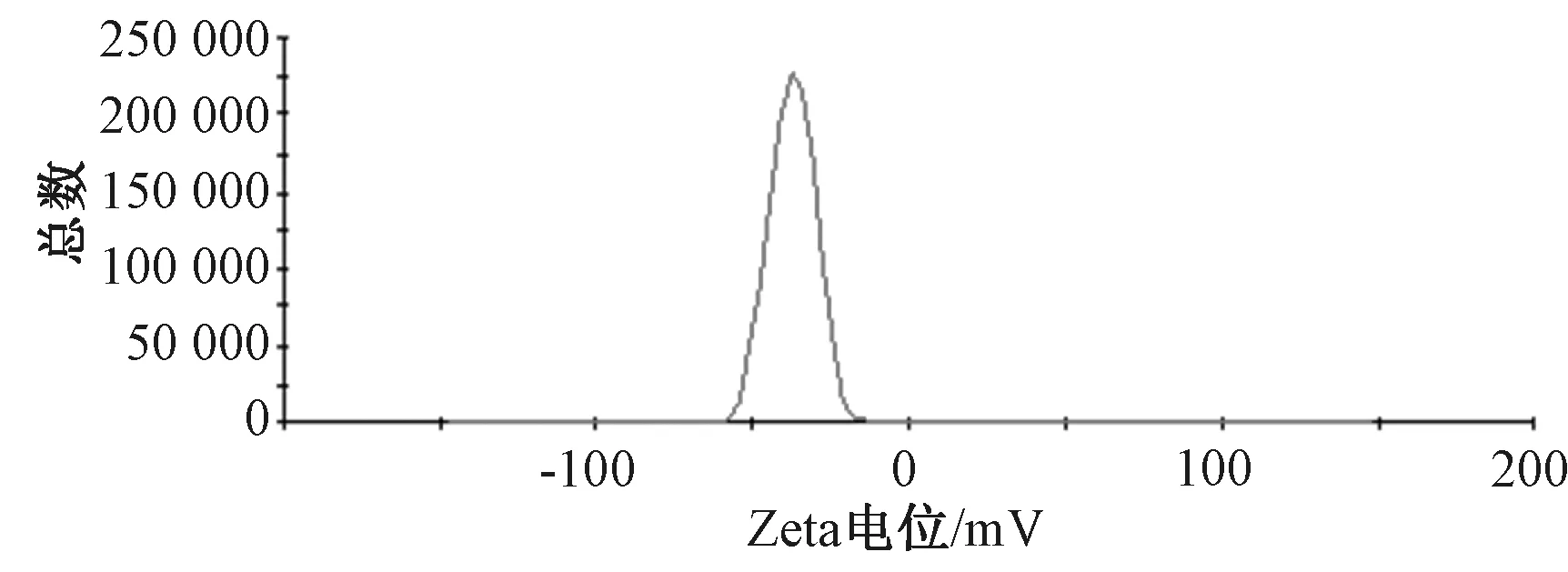
圖5 納米混懸劑Zeta電位Fig.5 Zeta potential of nanosuspensions

圖6 納米混懸劑粒徑分布Fig.6 Particle size distribution of nanosuspensions
2.6 形態觀察 取納米混懸劑0.5 mL,20 mL蒸餾水稀釋后滴于銅網上,靜置3 min,濾紙吸去多余液體,滴加1.5%磷鎢酸,自然晾干,在透射電鏡下觀察其形態,結果見圖7。由此可知,所得納米混懸劑呈球形,無粘連現象。

圖7 納米混懸劑透射電鏡圖Fig.7 Transmission electron microscopic image for nanosuspensions
2.7 凍干粉制備和穩定性考察 取納米混懸劑4 mL,加入5%乳糖-甘露醇(2∶3)混勻,在-60 ℃下預凍2 d,置于-25 ℃冷凍干燥機中抽真空,靜置2 d,即得凍干粉。取適量(西林瓶密封)至恒溫恒濕箱中,設置相對濕度為75%,溫度為40 ℃,于1、3、6、9、15、18、21、30 d測定粒徑、PDI,結果見表1,可知凍干粉放置30 d后粒徑仍小于200 nm,PDI仍小于0.2,表明其穩定性良好。
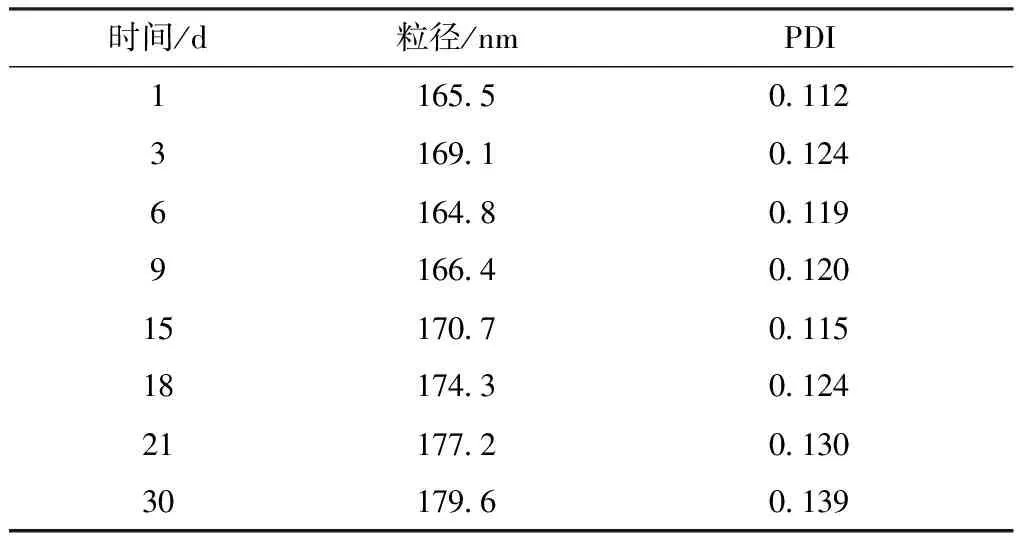
表1 穩定性試驗結果(n=3)
2.8 體外溶出度考察 配制1.5%十二烷基硫酸鈉,超聲處理10 min,作為溶出介質。取異甘草素、物理混合物凍干粉[取原料藥適量,再按照處方比例稱取輔料,蒸餾水將兩者混勻,然后加入5%乳糖-甘露醇(2∶3),在-60 ℃下預凍2 d后置于-25 ℃冷凍干燥機中抽真空,靜置2 d,即得]、納米混懸劑凍干粉適量(均過80目篩,異甘草素含量均為10 mg),加入2 mL溶出介質振蕩后置于透析袋中,兩端扎緊,在溶出介質體積900 mL、溫度(37±1)℃、轉速100 r/min條件下于0、5、10、15、30、45、60、75、90、105、120、150、180 min各取樣3 mL,補加1.5%十二烷基硫酸鈉,保持總溶出介質體積不變,0.22 μm微孔濾膜過濾,在“2.1.1”項色譜條件下進樣測定,計算累積溶出度,結果見圖8。由此可知,異甘草素、物理混合物凍干粉在180 min內的累積溶出度分別為39.71%、51.94%,后者更高的原因可能與輔料增溶作用有關[14];納米混懸劑凍干粉在45 min內累積溶出度為90.37%,在60 min內基本溶出完全。

圖8 異甘草素體外釋放曲線(n=3)Fig.8 In vitro release curves for isoliquiritigenin (n=3)
2.9 體內藥動學研究
2.9.1 分組與給藥 取異甘草素、物理混合物凍干粉、異甘草素納米混懸劑凍干粉適量,加入0.5%CMC-Na溶液使異甘草素質量濃度為10 mg/mL,作為灌胃藥液。將18只大鼠隨機分為異甘草素組、物理混合物凍干粉組、納米混懸劑凍干粉組,每組6只,禁食過夜,按照50 mg/kg劑量灌胃給予相應藥物,于0、0.5、0.75、1、1.5、2、3、4、6、8、10、12 h眼眶靜脈叢取血各約0.3 mL,置于肝素浸潤離心管中,2 500 r/min離心2 min,取上層血漿,置于-10 ℃冰箱中。
2.9.2 內標溶液制備 稱取苯甲酸鈉對照品10 mg至100 mL量瓶中,甲醇溶解定容并稀釋至200 ng/mL,即得。
2.9.3 血漿樣品處理 取大鼠血漿、內標溶液各100 μL,置于具塞尖低離心管中,加入1.5 mL甲醇,渦旋5 min后5 000 r/min離心10 min,取上清液,40 ℃氮氣緩慢吹干,加入100 μL甲醇復溶。
2.9.4 線性關系考察 取異甘草素對照品適量,甲醇制成質量濃度為1 800、900、600、200、100、20 ng/mL的溶液,精密量取100 μL,40 ℃氮氣緩慢吹干,加入空白血漿、內標溶液各100 μL,置于具塞尖低離心管中,作為血漿對照品溶液,按“2.9.3”項下方法處理,在“2.1.1”項色譜條件下進樣測定。以異甘草素、內標峰面積比值為縱坐標(Y),異甘草素質量濃度為橫坐標(X)進行回歸,得方程為Y=1.914 3X-0.587 6(r=0.993 5),在20~1 800 ng/mL范圍內線性關系良好。
2.9.5 方法學考察 取按“2.9.3”項下方法處理的血漿樣品,在“2.1.1”項色譜條件下進樣測定6次,測得異甘草素、內標峰面積比值的RSD為1.63%,表明儀器精密度良好。于0、4、8、12、24、48 h在“2.1.1”項色譜條件下進樣測定,測得異甘草素、內標峰面積比值的RSD為2.64%,表明樣品在48 h內穩定性良好。取“2.9.4”項下100、600、900 ng/mL對照品溶液,制成血漿對照品溶液,在“2.1.1”項色譜條件下進樣測定,測得異甘草素平均加樣回收率分別為89.37%、94.175、90.86%, RSD分別為5.82%、3.19%、4.97%。取25 ng/mL血漿對照品溶液,空白血漿逐步稀釋,按“2.9.3”項下方法處理,在“2.1.1”項色譜條件下進樣測定,分別以S/N=3、S/N=10為檢測限、定量限,測得兩者分別為2.5、6.0 ng/mL。
2.9.6 體內藥動學研究 異甘草素血藥濃度-時間曲線見圖9,其主要藥動學參數見表2。由此可知,物理混合物、原料藥tmax、Cmax、AUC0~t、AUC0~∞無顯著差異(P>0.05),表明前者促吸收作用較弱;與原料藥、物理混合物比較,納米混懸劑tmax縮短(P<0.01),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相對生物利用度增加至3.04倍。
3 討論
為了得到穩定的異甘草素納米混懸劑,選擇合適的穩定劑至關重要,它可阻止納米粒聚集,降低其碰撞幾率,抑制其生長。課題組前期對聚山梨酯80、聚乙烯醇、磷脂進行了篩選,但所得納米混懸劑的粒徑或PDI較大,而磷脂存在穩定性問題,最終選擇PVP K30、泊洛沙姆188作為穩定劑,其中泊洛沙姆188可吸附在納米粒子表面,其分子鏈具有舒展性,能阻止納米粒子在空間上彼此靠近[15];PVP K30可置換粒子表面OH-或H2O,產生吸附層,降低表面自由能,并且其分子鏈具有延展性,有助于防止納米粒子聚集[16],同時,上述聯合穩定劑也可提高溶出速率。

圖9 異甘草素血藥濃度-時間曲線(n=6)Fig.9 Plasma concentration-time curves for isoliquiritigenin (n=6)
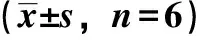
表2 異甘草素主要藥動學參數
藥動學研究結果表明,異甘草素、物理混合物Cmax、AUC0~t有所提高,可能是穩定劑具有增溶作用而促進吸收,但差異無統計學意義(P>0.05),而納米混懸劑tmax顯著縮短,可能是由于它在前30 min溶出速率較大,累積溶出度較高;Cmax、AUC0~t、AUC0~∞顯著升高,相對生物利用度增加至3.04倍,可能是由于納米藥物容易附著在腸道黏膜[17],并且它在伊派爾結中聚集,通過淋巴循循環進入血液循環而改變原料藥吸收途徑,使后者被高效吸收。
