家族性肌萎縮側索硬化癥一家系兩例
2022-06-27 07:50:22章軍建
醫學新知 2022年3期
關鍵詞:基因突變
李 曾,章軍建
武漢大學中南醫院神經內科(武漢 430071)
隨著遺傳學研究的進展,越來越多的肌萎縮側索硬化癥(amyotrophic lateral sclerosis,ALS)被證明與基因突變相關。不同的基因有不同的臨床表型,了解這些致病基因有助于疾病的精確診斷、預后判斷等。本文報告了兩例家族性ALS患者,均為SOD1基因p.Gly148Asp突變,癥狀在一年內快速進展,危及生命。本文對其臨床表現和遺傳評估進行分析,有助于加深對ALS的認識。
1 臨床資料
先證者,女,28歲。于2016年出現無明顯誘因的左下肢與右上肢無力、言語不清、飲水嗆咳、吞咽困難,無大小便障礙、肢體麻木等。家族中無近親結婚史及神經肌肉疾病家族史。查體見延髓麻痹、呼吸肌無力,四肢及軀干肌肉萎縮,四肢遠端肌力4-級、近端肌力4+級;四肢腱反射活躍,雙下肢巴賓斯基征陽性,余無明顯異常。肌電圖見高波幅、寬時限的運動電位,累及球段、頸段、胸段、腰段,提示廣泛神經源性病變。基因檢測提示SOD1基因存在c.443G>A雜合突變。完善頭部及脊髓MRI、腦脊液相關檢查,均未見明顯異常。排除其他疾病后,根據修訂版El Escorial診斷標準[1]考慮為擬診型ALS。后患者癥狀進行性加重,于2017年出現全身肌肉嚴重萎縮,咽喉肌和舌肌無力、萎縮,呼吸困難,以呼吸機維持通氣。發病2年后死于呼吸衰竭。
先證者母親,女,54歲。于2020年6月出現無明顯誘因左上肢無力,伴全身肉跳感,不伴感覺障礙。既往糖尿病病史、腸息肉手術史、女兒ALS病史,家族無近親結婚史。查體:體溫36.7℃,脈率79次/min,呼吸 20次/min,收縮壓/舒張壓111/68 mmHg(1 mmHg=0.133 kPa)。神志清楚,左上肢近端肌力3級、遠端肌力4+級,右上肢近端肌力5級、遠端肌力4+級,雙下肢近端肌力4級、遠端肌力5級。左側肩胛肌萎縮,左側肱二頭肌、雙側第一骨間肌肌容積減少,可見肌束震顫。雙上肢腱反射(++),雙下肢腱反射等稱降低,腹壁反射消失。無巴賓斯基征、霍夫曼征等病理反射征。其他神經系統評估包括認知、顱神經功能、感覺系統、言語和吞咽
均未發現異常。血常規、血生化、血脂、風濕免疫全套、微量元素、腫瘤全套未見明顯異常。腦脊液蛋白輕度增高(0.476 g/L),腦脊液壓力、常規、免疫、細菌檢查未見明顯異常。頸椎平掃與臂叢神經MRI檢查提示頸椎退行性病變,頸椎間盤(C3/4、C4/5、C5/6)稍突出。頭部彌散張量成像未見雙側皮質脊髓束明顯異常。肌電圖可見大的運動單位電位、纖維性顫動和正銳波的去神經支配電位,四肢周圍神經神經源性損害,主要累及運動髓鞘(包括球段、頸段、胸段、腰段)。肌電圖提示ALS或其他運動神經元疾病。獲患者知情同意后進行基因檢測,結果與先證者相同,SOD1基因存在c.443G>A雜合突變,導致其編碼氨基酸發生p.Gly148Asp錯義突變(圖1)。根據患者隱匿性起病,病程進行性加重,臨床癥狀如上、下運動神經元均受累并累及多個部位,肌電圖呈典型的神經源性損害,有陽性家族史,基因檢測提示SOD1基因突變,排除其他相關疾病,診斷為家族性肌萎縮側索硬化癥(familial amyotrophic lateral sclerosis,fALS)。發病2個月時ALS功能評分為38分。一年后隨訪,患者癥狀迅速進展,包括全身肌肉萎縮、構音障礙、吞咽與呼吸困難,于當地醫院重癥監護室內以呼吸機輔助通氣等維持生命,ALS功能評分下降到5分。
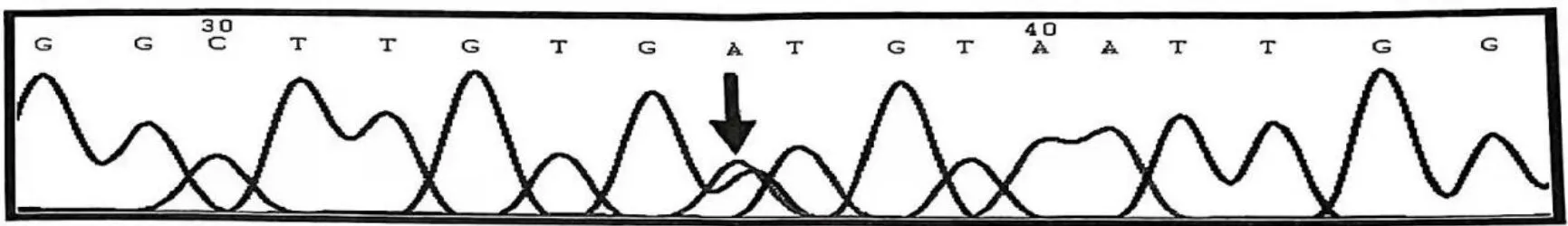
圖1 SOD1基因中c.443G>A雜合突變Figure 1. C.443G>A heterozygous mutation in SOD1 gene
2 討論
ALS是運動神經元病中一種致死性的神經退行性疾病,主要累及上、下運動神經元,導致進行性肌肉萎縮和痙攣,上肢遠端尤其突出。流行病學研究表明,ALS平均發病年齡為65歲,其中男性為55歲,女性為60歲,80歲以上人群發病率迅速下降,35歲前發病為早發性ALS[2]。ALS患病存在性別差異,男女患病比例約為1.3∶1。絕大多數病例為散發性ALS(sporadic amyotrophic lateral sclerosis,sALS),但仍有約5%~10%的患者為fALS,呈常染色體顯性或隱性遺傳模式。該病為節段性起病,隨著病情的發展,患者逐漸出現典型的上下運動神經元損害體征,表現為嚴重且廣泛的全身肌肉萎縮、肌張力增高、病理征陽性,并在發病3~5年后死于呼吸衰竭或肺部感染[3]。神經肌肉電生理改變主要表現為廣泛的神經源性損害,通常累及3個及以上區域(延髓、頸髓、胸髓、腰骶髓)。頭部及脊髓MRI無特征病灶。目前臨床上主要應用修訂版El Escorial標準診斷ALS,其根據患者的臨床表現、體征、病變累及范圍和肌電圖分為確診型、擬診型、實驗室支持的擬診型及可能型四個等級。對于fALS的診斷除依賴上述檢查手段外還需結合家族史、基因檢測等。該病應與多灶性運動神經病、脊髓空洞癥、脊髓型頸椎病、平山病、ALS綜合征相區別。本文討論的兩名患者均以肢體遠端無力起病,病程進展迅速,一年內累及球部、頸髓、腰髓三個部位,肢體及軀干肌肌肉萎縮,下運動神經元受損嚴重。上運動神經元受損體征不明顯,僅表現為腱反射活躍、病理征陽性。肌電圖提示神經源性損害,累及三個區域。患者具有錐體束受損表現,可排除多灶性運動神經病。無節段性痛溫覺缺失、無感覺障礙,可排除脊髓空洞癥及脊髓性頸椎病。患者無物理因素影響、無代謝異常、無免疫異常、無伴發腫瘤,可排除ALS綜合征。在一年期隨訪時,先證者母親已出現嚴重的咽喉肌萎縮、呼吸困難、肢體肌肉廣泛萎縮,結合兩名患者的病程、體征、肌電圖、家族史、基因檢測結果,排除其他疾病, 最終診斷為SOD1基因突變的fALS。
目前發現與ALS有關的基因有上百種[4],但相關致病基因僅30余種。研究表明,約68%的fALS和11%的sALS與致病基因相關。約三分之一的fALS病例中未發現非特異性基因突變[5]。已發現的致病基因中,最常見的是SOD1、TARDBP、FUS和C90ORF72[6]。其中SOD1基因是于1993年發現的第一個ALS致病基因[7]。SOD1基因位于21號染色體短臂,負責編碼銅鋅超氧化物歧化酶(Cu/Zn-superoxide dismutase,Cu/Zn-SOD)。Cu/Zn-SOD是細胞內清除自由基和抗氧化的重要酶,在將高反應性活性氧超氧化物轉化為氧分子和過氧化氫方面發揮著重要作用[8]。SOD1基因突變在ALS病例中占比較高,約25%的fALS和部分sALS均與其相關[9]。目前已經發現了SOD1基因180多種不同的突變,經典的突變包括p.D90A、p.A4V、p.G93A等。大部分突變為錯義點突變,呈常染色體顯性遺傳模式,它們并非聚集在基因的特定部分,而是散布在整個編碼區,且多發生在外顯子上,從而導致SOD1編碼蛋白的功能失常。SOD1基因突變相關的ALS患者病程進展緩慢,發病年齡較早,以下運動神經元體征為主,較少出現認知障礙[10]。但SOD1基因不同位點的突變會導致患者臨床特點、發病年齡、病程發展速度及生存期長短等有所不同。如攜帶p.A4V、p.H43R、p.L84V、p.G85R、p.N86S和 p.G93A等突變的患者,疾病往往進展迅速,平均生存期小于3年;而攜帶p.G93C、p.D90A或p.H46R等突變患者,以下運動神經元損害為主要表現,進展緩慢,生存期可達10年或更長。不同的突變方式臨床表現差異顯著,例如本文中兩例患者的基因突變類型均是將SOD1蛋白的148號密碼子上的甘氨酸替換為天冬氨酸,為G147D突變。該突變類型在國外fALS病例中已被報道,但尚未有關于此突變臨床特點的闡述。本文中兩名患者的臨床表現類似,不同于大多數的SOD1突變表型,其病程進展迅速,均在發病一年內嚴重累及呼吸肌和全身肌肉。一定程度上說明,該位點突變導致的臨床癥狀較一般SOD1突變更為嚴重,病程進展較快,以下運動神經元受損為主,引起全身肌肉特別是呼吸肌萎縮,且存活時間更短。但本文中兩例患者發病年齡存在差異,即使為同一基因相同位點的突變,其臨床表型,如臨床癥狀、發病年齡、病程的進展速度等也有一定的家族間或家族內的顯著區別。
ALS的治療方式包括藥物干預以及呼吸管理、營養支持、生活護理等綜合治療,一定程度上可延長患者的生存時間,提高患者的生活質量。利魯唑與依達拉奉聯用是目前唯一被批準治療ALS的藥物[11],但其只能延緩部分患者的病情進展,不能逆轉疾病。迄今為止,ALS尚無根治的方法[12]。相關治療的研究方向主要包括抗興奮毒性藥物、抗氧化和自由基清除劑、神經營養因子、基因治療等。現階段對于基因治療的研究較多,部分動物實驗已初顯成效,如在由SOD1編碼基因突變導致的fALS動物模型中,抑制突變基因可以提高存活率[13-14]。分子遺傳學研究發現遺傳因素在ALS疾病發生發展中起了重要作用。ALS-SOD1突變基因在脊髓運動神經元和中間神經元表達異常標志物,因此成為潛在的臨床靶標。Bravo-Hernandez等研究顯示在某些ALS發病前,軟脊膜下注射AAV9-shRNA-SOD1點基因治療,能顯著延長存活時間,大幅度保留運動神經元功能和運動能力,甚至在疾病發生后使用,也能阻止病程進展[15-16]。
隨著研究的深入,越來越多的ALS致病及風險基因被發現,相關基因治療臨床試驗的開展,促進了基因診斷應用于ALS臨床實踐的必要性。深入了解這些基因型對應的臨床表型,有助于疾病的精準診斷、預后判斷,并為今后的基因治療提供依據。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
