Crouzon syndrome in a fraternal twin: A case report and review of the literature
2022-06-28 04:15:34XiaoJingLiJiMeiSuXiaoWeiYe
World Journal of Clinical Cases 2022年16期
INTRODUCTION
Crouzon syndrome (CS) is an autosomal dominant inherited disorder and characterized by calvarial deformities with craniofacial dysostosis, exophthalmos, and facial anomalies[1,2]. In 1912, the French physician Louis Edouard Octave Crouzon first reported this syndrome and identified a mother and her son with craniosynostosis that resulted in skull and facial deformities[3]. This syndrome is described as the mildest type of many craniosynostosis syndromes[3,4]. The birth incidence of CS is approximately 1:25000[4]. It is proven to be the most common craniosynostosis syndrome, as it accounts for nearly 4.8% of all craniosynostosis cases at birth[1,5]. It is more common in males than females (3:1)[6]. Mutations in the() gene on chromosome locus 10q25.3-q26 are related to CS[7]. Families with CS commonly suggest an autosomal dominant inheritance pattern[8,9]. The syndrome presents incomplete penetrance and variable expressivity leading to phenotypical characteristics varying from mild to severe[7,10]. About 50% of cases result from de novo mutations of thegene[11].
在風險管控上,臺州市食品藥品監管局探索形成藥品生產“四環”工作法、醫療器械生產“ACE”工作法和藥品不良反應監測“123”工作法等風險管控機制,先后在全省會議和全國會議上作典型經驗介紹。而其實施的檢測資源市區一體化改革,縣級食品檢測資源整合試點經驗也獲得國家食品藥品監督管理總局的推廣,檢測資源整合績效評價體系研究課題成為全國樣本,市縣綜合檢測能力居全省前列。
CS is commonly observed at birth owing to characteristic facial and cranial deformities and a positive family history. The prominent manifestations of these patients can be detected upon physical examination. Brachycephaly, hypertelorism, proptosis, flattened forehead, beaked noses, and maxillary/midface hypoplasia are the most common features[3,12-14]. Hearing loss, strabismus (misaligned eyes), a cleft lip and palate, and dental problems can be also seen from these patients[12,15,16]. However, Crouzon syndrome patients have normal hands and feet in contrast to Apert syndrome patients and other craniosynostosis syndromes[17,18]. The different characteristics of the more common syndromic craniosynostosis syndromes are shown in Table 1[17,19-22].
The most common dental problems in craniosynostosis are supernumerary teeth, hypodontia, delayed eruption, and macrodontia[23,24]. We report a 6-year-old fraternal twin boy with CS who had many caries and enamel hypomineralization in the oral cavity. The boy’s parents and his fraternal twin sister did not show any abnormalities indicating CS. Therefore, we hypothesize that the twin boy’s gene mutation arose from a de novo mutation.
CASE PRESENTATION
Chief complaints
Li XJ and Su JM performed the initial review and data collection, reviewed the literature and contributed to the manuscript drafting and revision; Ye XW analyzed and interpreted the imaging findings; all authors issued final approval for the version to be submitted.
“否”在《齊》中未見,“否”在《周》中僅4見,位于句末作否定副詞,此用法在《國語》中已經出現,“‘否’是一個用法較特殊的否定副詞:它不作狀語,用于肯定否定迭用的句子中,表示否定的一面。”[5]《戰國策》中“否”也是此用法。例2“否”位于疑問句末時省略謂語中心,可補全。
History of present illness
A comprehensive treatment plan was made. The initial treatment involved improving the patient’s oral hygiene and restoring his carious teeth. The dental caries in teeth 54, 55, 64, 65, 74, 75, 85, 36, and 46 were treated with resin-based restorations. Topical fluoride was regularly applied to enamel hypomineralization in teeth 11, 21, 36, and 46 to promote enamel remineralization. The Skeletal Class III malocclusion was treated by orthodontic treatment and orthognathic surgery. Multidisciplinary approaches involving pediatric dentists, dental surgeons, oral and maxillofacial surgeons, and orthodontists were indispensable for this treatment.
History of past illness
The patient underwent intracranial decompression surgery at the age of 1 due to hydrocephalus. Posterior cranial vault distraction was performed when he was 2-years-old. The patient underwent a ventriculoperitoneal shunt operation for hydrocephalus at the age of 3. One year later, the patient had cranial vault remodeling as a result of craniofacial dysostosis.
In summary, CS could occur in a fraternal twin caused by a de novo mutation of thegene and is characterized by craniosynostosis, a prominent forehead, midface hypoplasia, and proptosis. Oral hygiene instruction and preventive programs on oral hygiene should be performed regularly. A multidisciplinary approach involving oral and maxillofacial surgeons and orthodontists was necessary for the treatment of midface hypoplasia.
Personal and family history
His medical and developmental history showed that he was born at full-term by cesarean delivery after an uneventful pregnancy with a 48 cm height and 2.35 kg weight, along with his fraternal twin sister with a 48 cm height and 1.8 kg weight. The family history of going back for three generations was negative for similar conditions, and his fraternal twin sister had no medical issues.
Physical examination
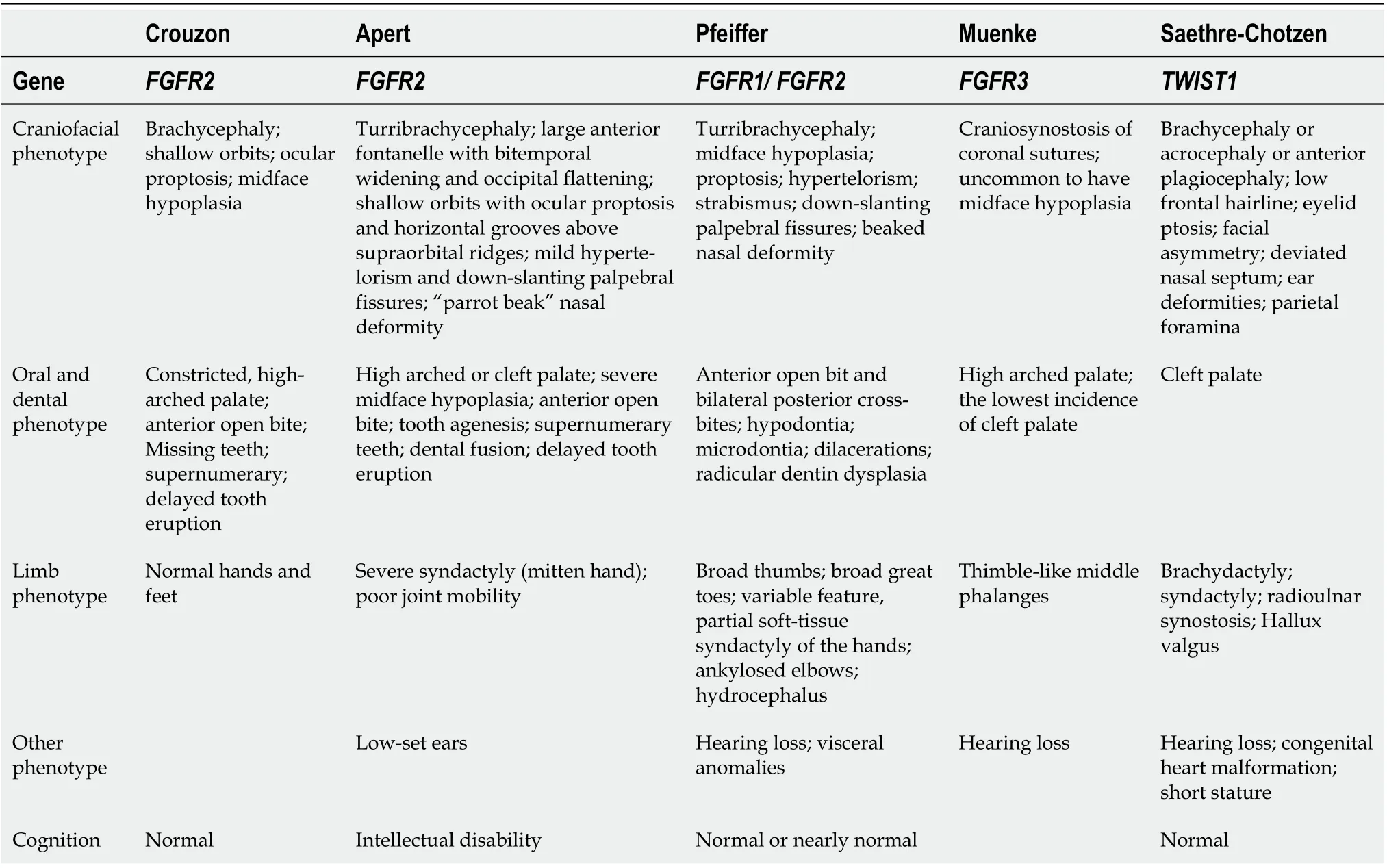
Physical examination showed normal height (108 cm) and weight (20.4 kg) compared with that of children of the same age. Clinically normal hands and feet were found. Extraoral examination revealed a prominent forehead, eyelid ptosis, ocular proptosis, shallow eye sockets, hypertelorism, midface hypoplasia, and a retrusive upper lip and protrusive lower lip (Figure 1).
Intraoral examination showed that the patient was in early mixed dentition, with all deciduous canines and molar teeth present in the maxillary and mandibular arch. The chronology of eruption and eruption status was in accordance with the child’s age (Figure 1). The oral hygiene of the child was poor with scattered pigmentation on the teeth, and permanent teeth 36 and 46 and deciduous teeth 54, 55, 65, 74, 75, and 85 were mostly decayed. The eruptive permanent teeth, including 11, 21, 36, and 46, showed enamel hypomineralization. The maxillary arch was constricted, and the anterior cross bite and open bite are shown.
黃驊,這座渤海岸邊的明珠一樣的城市,在一片生機盎然的沃土上,美麗鄉村建設使這里天藍、地綠、路潔、水清……如今,目光企及的地方,那些童年的歌謠依舊在田野深處交響,希望的果實在陽光下四季的變遷中靜靜地釀造成醇美的酒,從最南端的小堤柳莊村到最北端的東聚館村,那些美麗的鄉村建設得如一首田園詩,如一幅多彩的風情畫,如一串絕美的歌謠。
Laboratory examinations
Sanger sequencing showed that the pathogenic variant c.1026C>G (p.Cys342Trp) was present on thegene for the patientThe mutation was not present in any of the family members.
ESR檢測:空腹采集患者靜脈血,1.6 mL靜脈血加入到0.4 mL含109 mmol/L枸櫞酸鈉溶液的真空管中,混合均勻后放入Monitor-100型自動紅細胞沉降率分析儀(美國Monitor公司產品)的檢測位,靜置30 min后自動報告結果。正常值參考范圍:1~20 mm/h。
Imaging examinations
The final diagnosis of the presented case was CS, caries, and enamel hypomineralization.
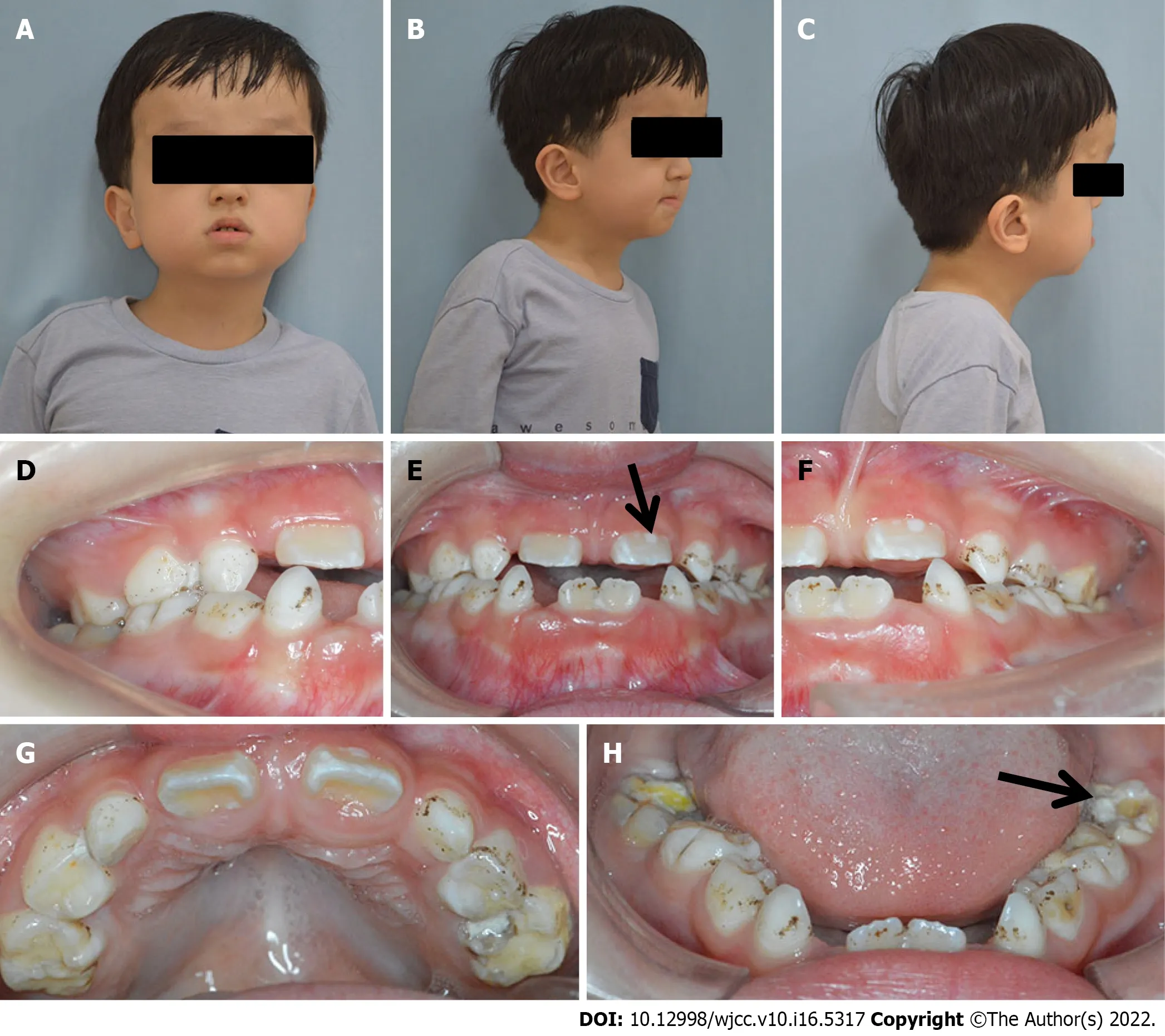
Based on the lateral cephalometric analysis, a severe skeletal Class III relationship was diagnosed with Sella-Nasion-A point angle: 63.8°, Sella-Nasion-B point angle: 71.6°, and A point-Nasion-B point angle: -7.8° (Figure 2B). A hypodivergent facial pattern was shown with Sella-Nasion/Gonion-Gnathion: 35.1°. The cephalometric analysis also revealed the maxillary skeletal size (condylion to point A) was diminished. The upper central incisors leaned toward the lip (U1/Sella-Nasion: 112.8°).
FINAL DIAGNOSIS
The panoramic radiograph showed that there were no supernumerary teeth or normal permanent tooth germ development. Deciduous carious teeth 55, 64, 65, 75, and 85 had once been restored and displayed further second caries at the time of the study. Permanent teeth 36 and 46 and deciduous teeth 54 and 74 were decayed (Figure 2A).
TREATMENT
Dental caries were observed since the patient was 4-years-old and gradually became progressively larger.
The authors declare that they have no conflict of interest.
OUTCOME AND FOLLOW-UP
Three months later, the oral hygiene of the patient was good, and topical fluoride was applied in teeth 11, 21, 36, and 46 to promote enamel remineralization.
DISCUSSION
Craniosynostosis results from the premature fusion of one or multiple cranial sutures, resulting in restricted growth of the skull, brain, face, and central nervous system development[4]. CS is the most common syndromic craniosynostosis and is caused by the mutation of[17].belongs to a family of fours. The FGFR family plays a primary role in the growth and differentiation of mesenchymal and neuroectodermal cells by binding to FGF and initiating signal transduction. Additionally, thefamily regulates cranial suture fusion on a macroscopic level[7].
For our patient the c.1026C>G (p.Cys342Trp) mutation on thegene caused a cysteine-totryptophan substitution at amino acid 342. Mutations at amino acid 342 can cause CS and Pfeiffer syndrome. Normal hands and feet can be observed with CS, which differentiates it from Pfeiffer syndrome. The loss of this cysteine residue is one of the most common mutations in CS patients and has previously been reported in a Chinese family, a Japanese sporadic case, and 4 Caucasian cases[9,27-29]. The Chinese family and 1 Caucasian family with CS have a dominant inherited amino acid 342 mutation[9,27]. The Japanese case and the German patients with CS were sporadic[28,29]. Since the fraternal twin sister and the parents did not present gene mutations and similar presentations, our patient’s mutation was considered to be a de novo mutation. In 11 of the 21 families with CS who underwent tracing to determine the origin of the mutation by analyzing parents and the other family members,mutations arose de novo, representing a high mutation rate for[29].
This case report describes a patient with CS, who presented with a high forehead, ocular proptosis, shallow eye sockets, hypertelorism, palpebral ptosis, and a skeletal Class III relationship. The carious teeth were fully restored, and oral hygiene instruction was performed in the clinic under behavior management and guidance. The child was advised to follow-up regularly with preventive care for teeth. The boy was satisfied with his good oral hygiene. Interceptive orthodontics was contemplated. Midfacial advancement with a Le Fort III, Le Fort II plus zygomatic repositioning, monobloc, or facial bipartition is required for midface retrusion[30]. The appropriate time of surgery is deemed at 8 years of age[31]. He has been referred to the Department of Oral and Maxillofacial Surgery for orthognathic surgical procedures in the next few years. He has also been referred to the Departments of Neurosurgery, Ophthalmology, Otolaryngology, and Psychology to prevent complications.
CONCLUSION
李漁(1611-1680),浙江金華府蘭溪人,初名仙侶,后改名漁,字謫凡,號笠翁。他是明末清初時期著名的文學家、戲劇家、戲劇理論家、美學家,一生中創作了大量的文學作品,包括戲曲、小說、詩詞、曲賦等,其中成就最高的是戲曲,他的作品將擬話本小說與戲曲藝術巧妙地融合為一個整體,創作出《笠翁傳奇十種》《無聲戲》《十二樓》等優秀戲曲作品。
ACKNOWLEDGEMENTS
We sincerely thank the patient and his parents for their participation and permission to publish this paper.
FOOTNOTES
A 6-year-old Chinese boy was referred to the stomatology department of the Children’s Hospital, Zhejiang University School of Medicine, with a chief complaint of dental caries.
Informed written consent was obtained from the patient for the publication of this report and any accompanying images.
At the first dental visit, the patient was nervous and fearful and was unwilling to open his mouth. Appropriately administered behavior management and guidance[25,26], including show-and-tell tactics, positive reinforcement, sound communication, and modeling, were used to manage the patient’s anxieties. Oral examination, radiographic assessment, oral prophylaxis, and polishing of the teeth with the rubber cup were performed during the first dental appointment. For the latter dental appointments, carious restoration was successfully completed utilizing show-and-tell tactics, positive reinforcement, and distracting the patient by showing movies during the procedure. After the procedure, the patient was praised and given a post-visit sticker, and he was agreeable for a future visit.
美國著名的圖書館學家達納先生早在1899年就曾說過,“一個圖書館的館藏,即使質量再好、貯藏再佳、排列再優,如果沒有好的館員,也是沒有什么價值的”[12]。
The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
China
Xiao-Jing Li 0000-0002-8122-9450; Ji-Mei Su 0000-0001-9429-617X; Xiao-Wei Ye 0000-0003-0518-5801.
2018年是ABB持續投資中國、優化業務布局的一年。例如,ABB在重慶設立的機器人應用中心正式開業,以重點滿足西部地區汽車、3C產品制造、裝備制造和消費品制造等領域對工業機器人快速增長的市場需求;不久前,ABB宣布將投資1.5億美元,在上海康橋建設其全球最大、最先進的機器人超級工廠,以擴大產能,提升效率,鞏固領先的市場地位,更好地滿足激增的客戶需求;而投資額約3億美元的ABB廈門工業中心昨天剛剛舉行揭幕儀式,從而將分散于廈門島內的多家企業合而為一,全面提升ABB自身的生產效率和核心競爭力。
Chen YL
Filipodia
完善的安全管理體系離不開資金的支持,施工單位應該加大在安全管理這一方面的資金投入,及時的對腳手架設備進行維護和更新。淘汰達到使用年限的設備或者是傳統性能落后的設備,使用性能優良,安全性能好的設備,這樣才能夠大大降低安全事故的發生率,保證施工工作順利進行,提高腳手架工程施工的質量。
Chen YL
猜你喜歡
當代陜西(2021年20期)2022-01-19 03:23:46
格言·校園版(2021年28期)2021-11-26 10:56:04
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
新世紀智能(高一語文)(2021年3期)2021-07-16 08:30:14
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
海峽姐妹(2018年9期)2018-10-17 01:42:44
戲曲研究(2017年3期)2018-01-23 02:50:39
 World Journal of Clinical Cases2022年16期
World Journal of Clinical Cases2022年16期
- World Journal of Clinical Cases的其它文章
- Practical points that gastrointestinal fellows should know in management of COVID-19
- Electroconvulsive therapy plays an irreplaceable role in treatment of major depressive disorder
- Pleural involvement in cryptococcal infection
- Advances in the clinical application of oxycodone in the perioperative period
- Endoscopic surgery for intraventricular hemorrhage: A comparative study and single center surgical experience
- Pediatric acute myeloid leukemia patients with i(17)(q10) mimicking acute promyelocytic leukemia: Two case reports