超高效液相色譜-串聯質譜法測定化妝品中莫西沙星等喹諾酮類禁用物質*
2022-06-30 10:01:12黨昊星曹海榮李曉宇
肥料與健康 2022年2期
關鍵詞:標準
黨昊星, 曹海榮, 李曉宇
(1.上海化工研究院有限公司 上海 200062;2.上海應用技術大學化學與環境工程學院 上海 201418)
0 前言
喹諾酮類藥物指的是人工合成的含4-喹諾酮結構的抗菌藥,具有抗菌譜廣、口服方便、價格低廉等特點,常見的喹諾酮類藥物有莫西沙星、雙氟沙星、沙拉沙星、氟羅沙星、氧氟沙星、恩諾沙星、培氟沙星、環丙沙星、依諾沙星、諾氟沙星等。由于喹諾酮類成本低,且具有一定的祛痘、除螨效果,生產廠商會在很多化妝品中添加一些喹諾酮類組分[1]。消費者如果錯用、誤用或過量使用含有喹諾酮類藥物的化妝品,很有可能引起過敏反應[2],嚴重時甚至有致癌的可能[3]。有些喹諾酮類藥物還被發現能使致病菌產生耐藥性[4],不利于臨床治療。因此,我國已在《化妝品安全技術規范》(2015版)中將喹諾酮類藥物列為化妝品禁用組分[5],研究有關化妝品中多種喹諾酮類藥物同時測定的方法非常必要。
目前,檢測化妝品中喹諾酮類藥物的常用方法有電化學法[6]、毛細管電泳技術[7]、表面增強拉曼技術[8]、高效液相色譜法[9-11]、液相色譜-質譜聯用法[12-14]等。文獻[1,15-16]報道用液相色譜-串聯質譜法或反相液相色譜法檢測化妝品中多種抗生素,檢測一個樣品所用時間超過20 min;文獻[3,10,12,17]報道的方法僅針對家禽畜肉中一種或多種喹諾酮類物質的檢測。由于上述檢測方法普遍存在操作煩瑣、耗時較長、適用范圍窄等不足,因此需要建立一種簡便、快速測定化妝品中多種喹諾酮類物質的方法。本文基于超高效液相色譜-串聯質譜法,提出了一種同時測定化妝品中莫西沙星等10種喹諾酮類藥物的分析方法,其中液相色譜-質譜聯用分析方法選擇性強、分辨率高、準確性好、靈敏度高[17],可以實現對10種喹諾酮類組分的有效分離及定性定量分析,并可應用于化妝品檢測。
1 試驗部分
1.1 儀器與試劑
ACQUITY UPLC?H-CLASS超高效液相色譜-Xevo TQD MS三重四極桿質譜聯用儀,美國Waters公司;Simplicity超純水純化系統,美國Millipore公司;KUDOS科導超聲波清洗器,上海科導超聲儀器公司;AL204-IC型分析天平,瑞士梅特勒-托利多公司。
莫西沙星(98.00%,質量分數,下同)、雙氟沙星(96.18%)、沙拉沙星(85.82%)、恩諾沙星(99.90%)、環丙沙星(92.31%)、培氟沙星(98.60%)、諾氟沙星(97.29%)、氧氟沙星(99.31%)、氟羅沙星(98.48%)、依諾沙星(95.84%)標準品,上海安譜實驗科技股份有限公司;甲醇、甲酸,色譜純,德國CNW Technologies。
混合標準儲備溶液:稱取莫西沙星、雙氟沙星、沙拉沙星、氟羅沙星、氧氟沙星、恩諾沙星、培氟沙星、環丙沙星、依諾沙星、諾氟沙星標準品各10 mg(精確至0.001 g)置于50 mL燒杯中,用甲醇溶解后轉移至100 mL容量瓶中,稀釋至刻度并搖勻,配制成100 mg/L的混合標準儲備溶液。
單標準儲備溶液:稱取莫西沙星、雙氟沙星、沙拉沙星、氟羅沙星、氧氟沙星、恩諾沙星、培氟沙星、環丙沙星、依諾沙星、諾氟沙星標準品各10 mg(精確至0.001 g)分別置于不同的50 mL燒杯中,用甲醇溶解后轉移至100 mL容量瓶中定容,搖勻,配制成100 mg/L的單標準儲備溶液。
1.2 儀器工作條件
1.2.1 色譜條件
ACQUITY UPLC BEH C18色譜柱(2.1 mm×50 mm,1.7 μm),柱溫為25 ℃,進樣量為5 μL;流動相中有機相選用甲醇(A),水相為0.2%(體積分數,下同)甲酸溶液(B),流量為0.3 mL/min,梯度洗脫程序見表1。
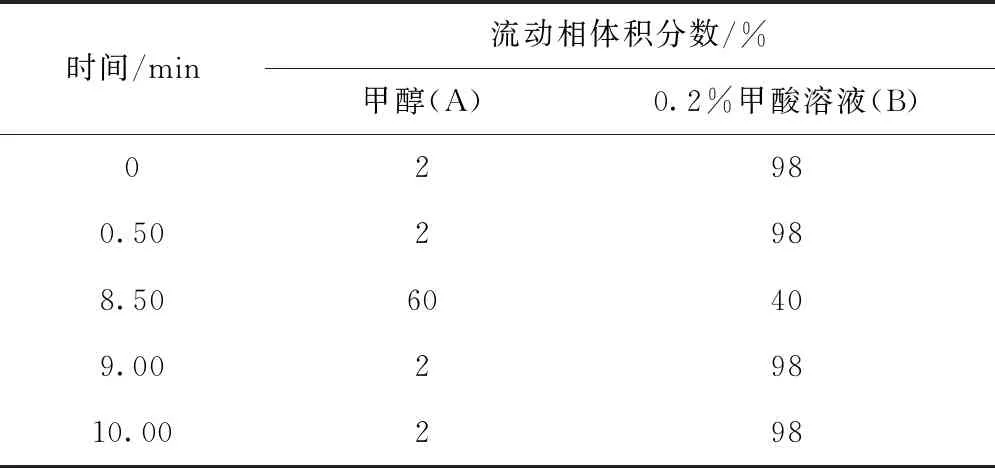
表1 梯度洗脫程序
1.2.2 質譜條件
電噴霧正離子源(ESI+)模式,多反應監測(MRM)模式,干燥氣溫度為600 ℃,干燥氣流量為900 L/h,錐孔流量為50 L/h,毛細管電壓為0.50 kV。
1.3 樣品前處理
稱取化妝水空白樣品1 g(精確至0.001 g)置于50 mL比色管中,加入2 mL飽和氯化鈉溶液和2%甲酸溶液30 mL,超聲5 min,加入甲醇10 mL,超聲振蕩15 min,靜置3 min后,將上層清液進行過濾,得到的濾液作為待測樣品。
2 結果與討論
2.1 質譜條件的優化
將已制備的單標準儲備溶液經蠕動泵注入到離子源中進行質譜分析后,得到目標物的母離子質荷比,通過對比正、負離子模式下的響應值發現,在ESI+模式下,喹諾酮類物質均有較強的分子-離子峰,因此試驗選用電噴霧正離子源模式。
采用ESI+進行二級質譜分析,通過調節碰撞能量(CE)和電壓參數,選擇并確定響應較好的定量離子和定性離子對,使待測物的離子化效率達到最優,同時確定最優的質譜條件。10種喹諾酮類分析物的質譜參數見表2。
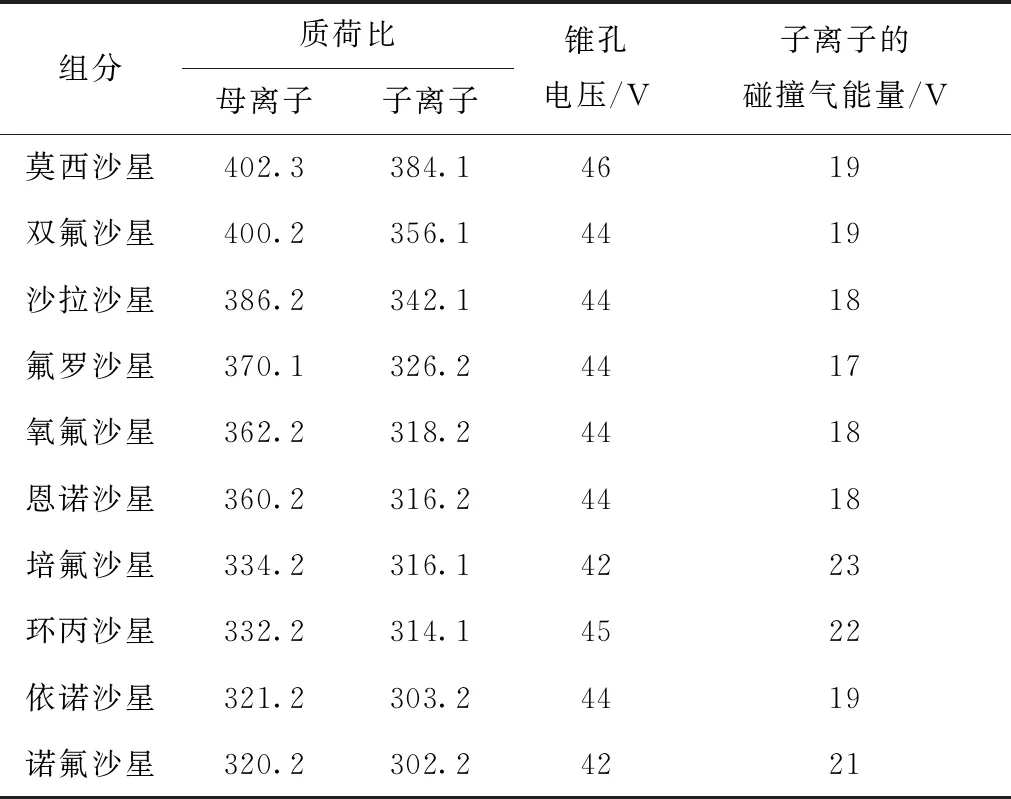
表2 10種喹諾酮類分析物的質譜參數
2.2 流動相的選擇
首先制備乙腈-0.2%甲酸溶液、乙腈-0.1%甲酸溶液、甲醇-0.1%甲酸溶液和甲醇-0.2%甲酸溶液等混合溶液,分別作為流動相;將10種喹諾酮類分析物溶于甲醇和乙腈,靜置搖勻后經色譜柱進行分析。結果表明,選用甲醇-0.2%甲酸溶液作為流動相時,峰形較明顯,各組分的分離度較高。因此,試驗選用甲醇-0.2%甲酸溶液作為流動相[18]。
考慮到氟羅沙星、培氟沙星等10種喹諾酮類分析物相互之間存在較為明顯的極性差異,選用梯度洗脫程序。在梯度洗脫條件下,10種喹諾酮類分析物的MRM色譜圖見圖1。
2.3 方法學驗證
2.3.1 線性關系、檢出限和定量限
(1)線性關系
移取10種標準物質的混合標準儲備溶液,用甲醇稀釋后得到5、10、20、50、80、100 μg/L混合標準系列溶液。將混合標準系列溶液按儀器工作條件分別進樣,以混合標準系列溶液的質量濃度為橫坐標,對應的峰面積為縱坐標繪制標準曲線。結果表明,10種喹諾酮類禁用物質的質量濃度在5~100 μg/L內與峰面積呈線性相關,相關系數為0.998 2~0.999 8。
(2)檢出限和定量限
記錄最低質量濃度待測樣品的信號和空白樣品信號(高度)的比值,以3倍信噪比和10倍信噪比分別計算檢出限LOD和定量限LOQ[19],計算公式見式(1)和式(2):
(1)
(2)
式中:Cmin——標準曲線中最低質量濃度;
S/N——信噪比。
計算得10種喹諾酮類禁用物質的檢出限為0.002 53~0.025 5 mg/kg,定量限為0.008 44~0.085 2 mg/kg。
10種喹諾酮類禁用物質的線性范圍、相關系數、檢出限及定量限見表3。
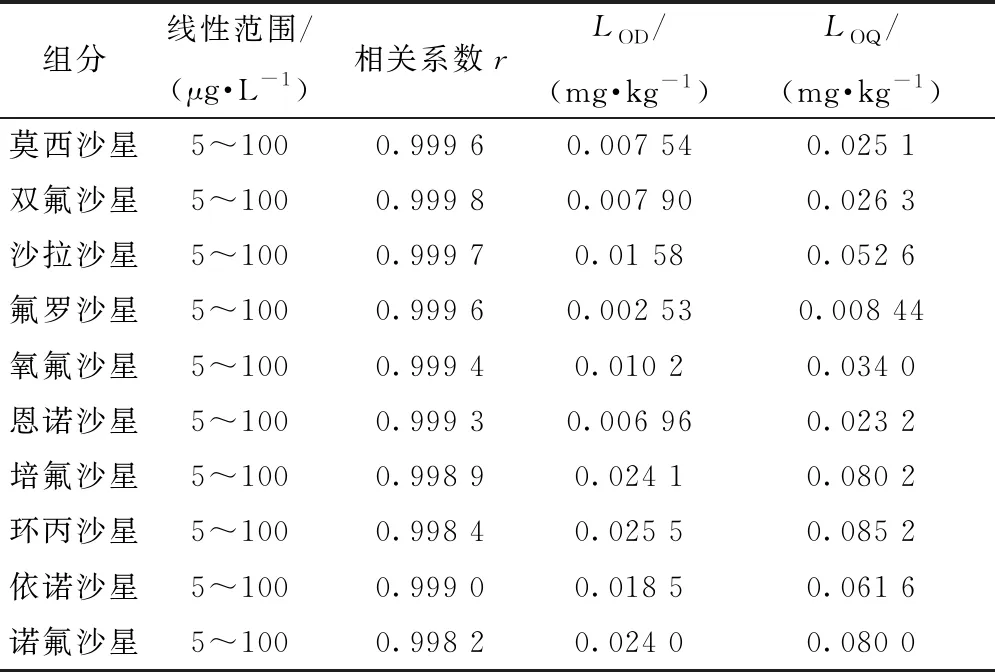
表3 10種喹諾酮類禁用物質的線性范圍、相關系數、檢出限和定量限
2.3.2 加標回收率
稱取1 g待測樣品,加入至混合標準儲備溶液中,按試驗方法處理后得到質量濃度分別為0、40、60、80 μg/L的4個不同樣品;采用選定的儀器工作條件,每個樣品進行3組平行測定,共測定12次,計算加標樣品的回收率,計算公式見式(3)[20]:
(3)
式中:ρi——第i次測量的質量濃度;
ρ0——待測樣品的質量濃度;
ρs——實際加入的混合標準儲備溶液的質量濃度。
以加入40 μg/L混合標準儲備溶液為例,在加標回收試驗過程中,空白樣品的MRM色譜圖和加標樣品的MRM色譜圖分別見圖2和圖3。
10種喹諾酮類禁用物質的加標回收率(見表4)為100.00%~116.43%,說明試驗方法準確度較好。
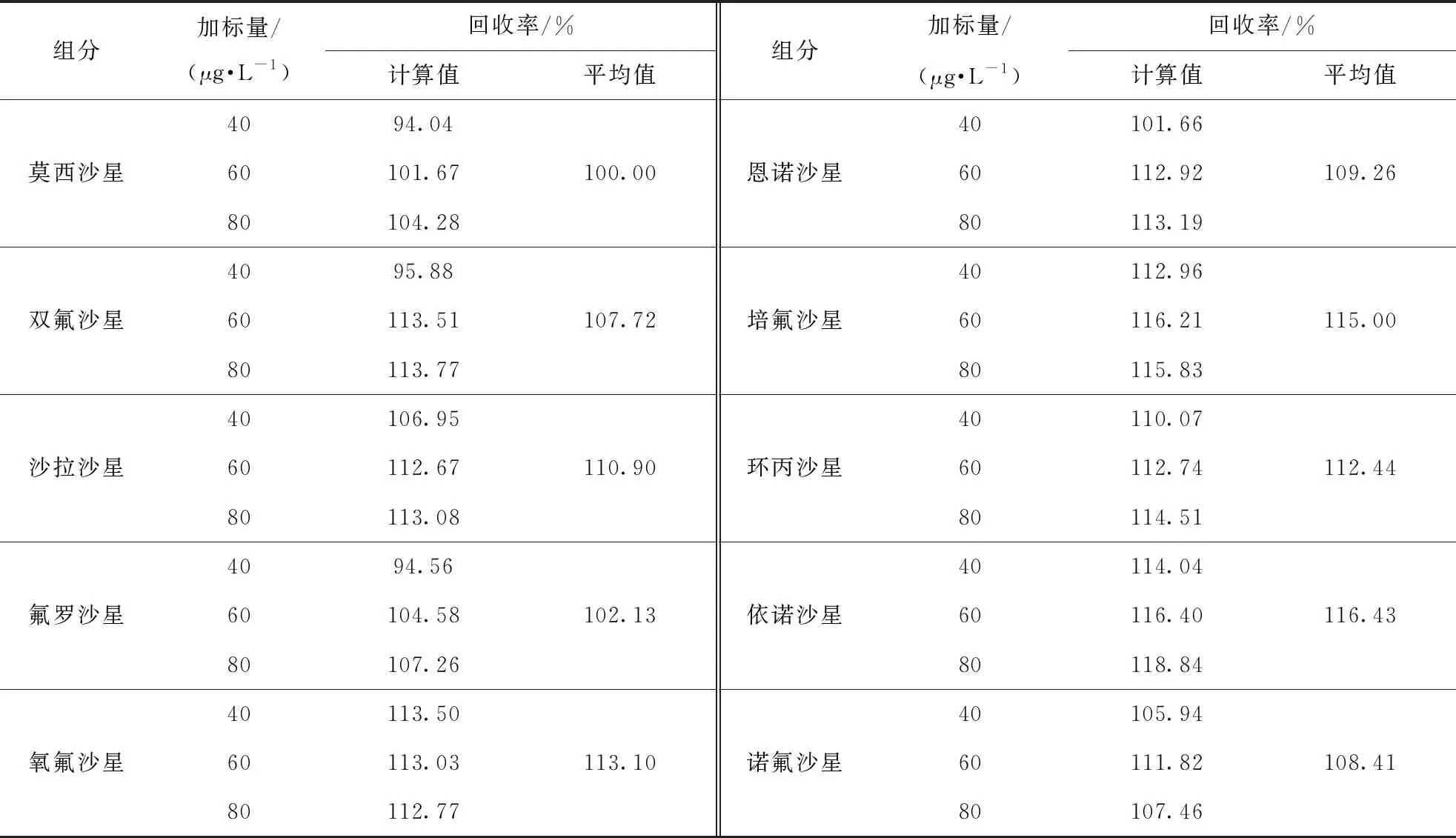
表4 10種喹諾酮類禁用物質的加標回收率
2.3.3 精密度
稱取1 g待測樣品,加入至混合標準儲備溶液中,得到質量濃度為40、60、80 μg/L的3個試樣;在選定的儀器工作條件下,每個樣品進行6次平行測定,計算測定值的相對標準偏差(RSD),計算公式見式(4)和式(5)[20]:
(4)
(5)
式中:S——質量濃度測量值的標準偏差;
ρi——第i次測定的質量濃度;
n——測定次數。
試驗測得的10種喹諾酮類禁用物質的RSD(見表5)為1.77%~5.90%(n=6),小于15%,表明試驗方法的精密度較好。
3 結語
本文提出了一種同時測定化妝品中莫西沙星、雙氟沙星、沙拉沙星、氟羅沙星、氧氟沙星、恩諾沙星、培氟沙星、環丙沙星、依諾沙星、諾氟沙星等10種喹諾酮類禁用物質的超高效液相色譜-串聯質譜法。方法簡便快捷、耗時較短,標準曲線的相關系數為0.998 2~0.999 8,10種物質的檢出限為0.002 53~0.025 5 mg/kg,定量限為0.008 44~0.085 2 mg/kg,加標回收率為100.00%~116.43%,精密度平均值小于15%,表明所提出方法的靈敏度較高、準確性較好、可重復性較好,可快速測定化妝品中多種喹諾酮類藥物,以期為化妝品中禁用物質的檢測提供技術支持。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
