食用油中多環芳烴檢測的前處理方法研究進展
2022-07-02 03:49:42孟祥茹胡樂乾尹春玲
食品科學 2022年11期
孟祥茹,胡樂乾*,琚 熒,張 艷,尹春玲*
(河南工業大學化學化工學院,河南 鄭州 450001)
食用油是人們膳食結構中的重要組成部分,可為人體提供必需脂肪酸、VE、植物甾醇等營養物質,在人們生活中發揮著重要的作用。然而近年來,食用油中多環芳烴的檢出多有報道,主要污染來源有種子的污染、油料加工的污染、油脂生產工藝上的污染以及食品外包裝的遷移污染。且有研究顯示隨著油脂煎炸時間的延長,多環芳烴及其衍生物的含量也有所增加。食用油脂是人體攝入多環芳烴的主要來源之一,以苯并[]芘為例,GB/T 2762ü2017《食品安全國家標準 食品中污染物限量》規定油脂及其制品中苯并[]芘最大限量為10 μg/kg。
多環芳烴是兩個或兩個以上的芳香族環的復雜化合物的總稱,主要是由石油、煤、天然氣、煙草、木料、煙熏食品和一些有機化合物熱解或不完全燃燒產生的,在食品、水、土壤、灰塵等中廣泛存在。多數多環芳烴不溶于水,易溶于有機溶劑,具有高親脂性、高熔點和高沸點,同時也具有較強的生物富集性。它們的化學性質穩定、不宜降解,是環境的主要污染源之一。多環芳烴根據苯環的數目可以分為兩類:輕質多環芳烴(4個或少于4個苯環)和重質多環芳烴(5個或更多苯環),通常重質多環芳烴要比輕質多環芳烴更穩定,毒性更強。研究表明,大多數的多環芳烴具有致癌性、致畸性和致突變性,它們可以通過身體脂肪迅速分布到人體各種組織中。其中,一些多環芳烴的代謝物具有與細胞蛋白質和DNA結合的能力,具有毒性作用,能夠對細胞造成損傷,從而導致突變和癌癥。長期食用多環芳烴含量高的食用油會對人體造成一定的危害,因此對食用油中的多環芳烴進行檢測十分重要,其檢測方法對于鑒定食用油品質具有重要影響。
食用油中多環芳烴的檢測主要包括樣品的前處理和測定兩個基本過程,其中常用的測定方法有氣相色譜法、高效液相色譜法、超高效液相食用油中多環芳烴色譜法、表面增強拉曼光譜法、熒光分光光度法、色譜聯用技術等。雖然檢測方法眾多,但檢測儀器對樣品要求較高,檢測前大多都需要對樣品進行前處理(除雜純化富集多環芳烴),由于食用油中脂肪含量高,脂質基質復雜,且多環芳烴具有高親脂性,前處理過程對于分析食用油中的多環芳烴是一個非常關鍵的過程,但目前仍然是一個很大的挑戰,因此尋找合適的預處理方法,開發快速準確的方法來檢測食用油中的多環芳烴具有重要意義。本文重點對近年來食用油中常用多環芳烴檢測的樣品前處理技術進行總結,并比較了不同前處理方法的優劣,以期為建立更高效的油脂中多環芳烴檢測的前處理方法提供參考。
1 食用油中多環芳烴的前處理技術
由于多環芳烴的親脂性高,在提取多環芳烴時,通常食用油中脂類成分會一起被提取出來,這會對食用油中痕量多環芳烴的檢測造成較大的干擾,對測定結果產生不利影響。為了更有效地提取和富集多環芳烴,同時去除甘油三酯、脂肪酸等脂類雜質,需要選擇合適的處理方法對油脂樣品進行預處理,達到消除基體干擾,提高檢測靈敏度的目的。目前,食用油中多環芳烴檢測的前處理方法可以分為分離和純化兩個過程,分離方法主要有皂化法、索氏提取法、液液萃取法、超聲波輔助萃取法、微波輔助萃取法;純化方法主要有柱層析法、固相萃取、固相微萃取、分散固相萃取法、磁性固相萃取、分子印跡萃取等。
1.1 分離方法
1.1.1 皂化法
皂化法是利用堿性物質與食品中的脂肪等雜質發生皂化反應,進而除去脂肪酸鹽的一種處理方法。由于多環芳烴是親脂性物質,采用皂化法可以除去較多的脂質,達到分離、純化多環芳烴的目的。常見的皂化法處理是采用氫氧化鉀的乙醇或甲醇溶液在回流條件下分離油脂樣品中的多環芳烴。
Zachara等采用皂化法,用1.5 mol/L的氫氧化鉀-甲醇溶液回流90 min皂化植物油,并用環己烷進一步萃取,經濃縮、氧化鋁柱凈化,采用高效液相色譜-熒光檢測器法(high performance liquid chromatography-fluorescence detection,HPLC-FLD)檢測植物油中的4種多環芳烴,檢出限為0.18 μg/kg,定量限為0.25 μg/kg,相關系數大于0.998,回收率為80%~110%。Akdo?an等采用皂化法,在60 ℃下用5 mol/L的氫氧化鉀-乙醇溶液回流60 min皂化植物油,并用甲苯進一步萃取,經濃縮、硅膠固相萃取柱凈化,采用HPLC-FLD檢測植物油中的4種多環芳烴,在最優條件下,決定系數為0.998 0~0.999 9,檢出限為0.06~0.12 μg/kg,定量限為0.13~0.24 μg/kg,回收率大于84.8%。Dost等采用皂化法,在60 ℃下用1 mol/L的氫氧化鈉的甲醇-甲苯(體積比為1∶1)的混合溶液回流60 min皂化植物油,用甲苯進一步萃取,經濃縮、硅鋁柱(質量比1∶1)凈化,采用HPLC-紫外-可見檢測器(ultraviolet-visible detector,UV-Vis)檢測植物油中的9種多環芳烴,決定系數為0.995 1~0.999 6,檢出限為0.26~1.15 μg/L,定量限為0.87~3.84 μg/L,回收率為80%~104%。此外,據報道一些學者認為苛刻的堿性處理可能會對樣品中不穩定的多環芳烴產生影響。
在傳統分離方法中一般會選擇皂化法作為初步的前處理步驟,之后選擇環己烷、二氯甲烷、,-二甲基甲酰胺(,-dimethylformamide,DMF)等溶劑進行進一步的液液萃取和純化,以便分離油脂中多環芳烴。皂化法可以有效地去除食用油中大部分脂質,該方法簡單廉價,適用于普通實驗室。但是有機溶劑消耗大、耗時長,且僅用皂化法萃取會有雜質殘留,必須進一步純化,導致整個前處理過程繁瑣耗時,不適合快速檢測。
1.1.2 索氏提取法
索氏提取法也稱作連續提取法,是一種傳統、經典的前處理方法。王麗霞等采用自動索氏提取法提取,硅膠固相萃取柱凈化,HPLC-UV-FLD法檢測油炸面制品中的16種多環芳烴,該方法的相關系數大于0.999 0,回收率為92.5%~106.4%,回收率較高,但是耗費時間較長。Sun Ying等采用索氏提取法提取,并分別采用分子印跡固相萃取法和凝膠滲透色譜法兩種方法凈化,通過氧化鋁固相萃取柱進一步純化,并用氣相色譜-質譜(gas chromatograph-mass spectrometer,GC-MS)法檢測五花肉、黃油以及人臍帶中的16種多環芳烴,結果表明分子印跡固相萃取法和凝膠滲透色譜法均能獲得良好的回收率,除萘的回收率約為50%,其余多環芳烴的回收率均為75%~120%,結果較好。
索氏提取法經典方便,不需要復雜的儀器設備,回收率較好,但是有機溶劑需求大,操作繁瑣、耗時,不適合快速檢測,近幾年使用較少。由此可見,開發快速、低成本、操作簡便、綠色環保的前處理檢測方法,并實現應用于油脂中多環芳烴的現場檢測,是目前的研究熱點。
1.1.3 液液萃取法
液液萃取是一種傳統的樣品前處理方法,利用目標分析物在兩種互不相溶的有機溶劑中溶解度不同,從而使目標分析物轉移到富集溶劑中,達到分離、富集目標分析物的目的。一般情況下,液液萃取需要進行多次萃取以提高回收率。食用油中多環芳烴傳統的前處理過程是直接進行液液萃取或者先皂化處理后再進行液液萃取,萃取結束后采用柱層析法進行純化,去除干擾物,富集多環芳烴。
Rascón等采用液液萃取法,將油樣用正己烷溶解,然后用DMF-水(體積比為9∶1)的混合溶液萃取,之后運用以C為吸附劑的半自動固相萃取系統進行純化,并利用GC-MS法測定橄欖油、芝麻油、大豆油等5種食用油中的16種多環芳烴,該方法的相關系數大于0.995,檢出限為0.004~0.110 μg/kg,定量限為0.012~0.330 μg/kg,回收率為87%~104%,該方法靈敏度、選擇性和精密度較高。Zhou Ruize等采用液液萃取法,將油樣用己烷飽和的乙腈溶液萃取,并通過GC-MS法檢測大豆油、花生油、橄欖油和玉米油中的多種環境污染物,其中23種多環芳烴的相關系數大于0.995,檢出限為0.1~1.0 μg/kg,回收率為70.0%~110.8%,該方法避免了大量油脂和色素的干擾。Camargo等采用液液萃取法,將大豆油用己烷溶解,然后用DMF-水(體積比為9∶1)混合溶液萃取,C固相萃取柱進行純化,并通過HPLC-FLD測定大豆油中的13種多環芳烴,該方法的決定系數大于0.999,檢出限為0.02~0.76 μg/kg,定量限為0.03~0.96 μg/kg,回收率為71%~115%。Molle等通過同樣的方法測定菜籽油、葵花籽油和玉米油中的13種多環芳烴,該方法的相關系數為0.993 3~0.998 9,檢出限為0.07~0.30 μg/kg,定量限為0.3 μg/kg,回收率為71%~110%。然而該分析方法對苯并[]熒蒽(benzo[]fluoranthene,BjF)和茚并[1,2,3-,]芘(indeno[1,2,3-,]pyrene,IcdP)的靈敏度較低,這2種多環芳烴的檢出限分別為1.95 μg/kg和1.32 μg/kg,定量限為3.0 μg/kg。Oh等采用液液萃取法,將油樣用乙腈液液萃取,弗羅里硅土固相萃取柱凈化,結合同位素稀釋(isotope dilution,ID)-GC-MS測定食用油中的鄰苯二甲酸鹽、己二酸鹽和多環芳烴,其中8種多環芳烴的決定系數為0.991 7~0.999 9,檢出限為0.15~0.77 μg/kg,定量限為0.44~2.33 μg/kg,回收率為80.6%~96.9%。
作為一種傳統的萃取方法,液液萃取的優點是操作簡單、成本低廉、提取效率較好,但是易乳化,導致兩相分離較慢,并且需要多次萃取分離來提高回收率,有機試劑消耗大,可能造成環境污染。綜上所述,隨著科學的發展以及人們對綠色化學的需求增加,選擇體積小的萃取液進行提取,縮短相分離時間和前處理時間,提高萃取效率,實現微型化是當前液液萃取方法的一個研究趨勢。
1.1.4 超聲波輔助萃取法
超聲波萃取法又稱為超聲輔助萃取法,其原理是通過超聲波的空化作用和熱作用,使多環芳烴快速有效地溶解到溶劑中。
Liu Yihong等以二甲基亞砜為溶劑進行超聲輔助法萃取,通過同步熒光法檢測植物油中的4種多環芳烴,該方法的檢出限為0.16~13.00 μg/kg,回收率為69.3%~113.0%,該方法方便快捷、靈敏度高。Taghvaee等以乙腈和丙酮的混合溶液為溶劑,采用改良低溫法和改良超聲輔助液液萃取法,通過HPLC-FLD檢測橄欖油和精煉果渣油中的15種多環芳烴。其中改良超聲輔助液液萃取法分析時間更短,決定系數大于0.992 9,檢出限為0.16~0.97 μg/kg,定量限為0.57~2.93 μg/kg,回收率為75%~111%。Wu Shimin等以正己烷為溶劑,利用DMF稀釋,并進行超聲輔助液液萃取,以弗羅里硅土固相萃取柱凈化,通過GC-MS檢測4種食用油中的16種多環芳烴,決定系數為0.998 0~0.999 9,回收率為70.11%~127.92%,該方法減少了樣品和溶劑使用量。Zhao Weijun等以乙腈和丙酮的混合溶液為溶劑進行超聲輔助液液萃取,以C固相萃取柱凈化,通過HPLC-二極管陣列檢測器(diode-array detector,DAD)-FLD檢測大豆油、葵花籽油、玉米油等6種食用油中的16種多環芳烴,決定系數為0.998 8~0.999 9,檢出限為0.01~2.35 μg/L,定量限為0.04~7.00 μg/L,回收率為57.3%~94.6%。
超聲波萃取法簡單、快速、便宜、溶劑用量少、萃取效率高,近幾年在油脂多環芳烴的分離中得到了極大的應用,但在超聲過程中易出現超聲盲區,且超聲時間過長,一些雜質會被共提出。因此,超聲波萃取法一般需要多種前處理方法聯用以去除共提雜質,同時應控制超聲提取的能量和時間。減少超聲盲區及避免共提取雜質的產生仍是該方法有待解決的問題。
1.1.5 微波輔助萃取法
微波輔助萃取法又稱為微波輔助溶劑萃取,利用微波加熱達到快速萃取樣品中目標物的目的。微波加熱的原理是基于偶極轉動和微波對偶極子和帶電分子或離子的離子傳導效應。目前微波輔助萃取法在多環芳烴的萃取應用中比較多,但是在食用油中的應用較少。
Mohammadi等采用微波輔助分散液液微萃取法,結合GC-MS檢測食用油中的14種多環芳烴,該方法具有快速、簡便、溶劑消耗低、靈敏度高等優點,決定系數為0.934 6~0.997 8,檢出限為0.2~2.7 μg/L,定量限為0.6~9.1 μg/L,回收率為84.4%~101.9%。Alarcón等采用微波輔助液液萃取,二氧化硅固相萃取柱凈化,結合熒光分光光度法進行檢測,采用偏最小二乘法(partial least squares regression,PLS)和展開偏最小二乘法(unfold partial least squares regression,U-PLS)算法預測初榨橄欖油和葵花籽油中7種重質多環芳烴的含量,該方法的相關系數大于0.994,檢出限為0.8~7.0 μg/kg,定量限為2.4~22 μg/kg,且基于三維光譜數據的U-PLS算法,獲得了最佳分析靈敏度、最低檢出限和定量限,并通過HPLC-FLD對該方法進行評價,結果顯示回收率為62%~84%;Alarcón等又采用相同的萃取方法,結合U-PLS/殘差雙線性(residual bilinearization,RBL)和平行因子分析(parallel factors analysis,PARAFAC)算法對三維熒光光譜進行分析,測定初榨橄欖油和葵花籽油中7種重質多環芳烴,其中U-PLS/RBL算法在此實驗中更具優勢,檢出限為0.07~2.00 μg/kg,并采用HPLC-FLD法對該算法預測結果進行了評價,結果良好。
微波萃取法具有快速、環保、可批量處理樣品、提取效率高等優點,但是不適用易揮發組分的提取,且能量過高可能會導致樣品中其他成分也被提取出,因此需控制微波提取的能量和時間。此外微波萃取法需與其他樣品前處理方法聯用,以去除共提取物干擾。
1.2 純化方法
1.2.1 柱層析法
柱層析法又稱為柱色譜法,根據樣品中各組分在固定相和流動相中的分配系數不同,經過多次分配,將不同組分分離。采用柱層析法分離純化油脂中的多環芳烴時,固定相一般為中性氧化鋁或者硅膠,流動相一般選擇極性較弱的有機溶劑,如乙酸乙酯、石油醚、環己烷等。
Hossain等采用乙腈-丙酮(體積比為6∶4)混合溶液進行液液萃取,經過超聲、離心、濃縮以及硅膠柱凈化后,應用GC-MS檢測大豆油、芥末油、椰子油中的8種多環芳烴,該方法的決定系數大于0.987,檢出限為1.9~3.1 μg/kg,回收率為56%~84%。Zachara等采用皂化法進行分離,經液液萃取、濃縮后,采用氧化鋁柱凈化,結合HPLC-FLD檢測植物油中的4種多環芳烴,相關系數大于0.998,檢出限為0.18 μg/kg,定量限為0.25 μg/kg,回收率為80%~110%。Dost等采用皂化法進行分離,經液液萃取、濃縮后,采用硅鋁層析柱(質量比為1∶1)凈化,結合HPLC-UV-Vis檢測植物油中的9種多環芳烴,決定系數為0.995 1~0.999 6,檢出限為0.26~1.15 μg/L,定量限為0.87~3.84 μg/L,回收率為80%~104%。
柱層析法成本低廉、操作簡單,但是耗時長、溶劑用量大、重復性較差,無法滿足綠色環保的要求,近年來已經被固相萃取法取代。
1.2.2 固相萃取法
固相萃取是在柱層析基礎上發展起來的一種樣品前處理方法,原理是利用固體吸附劑吸附液體樣品中的目標分析物,后續用洗脫液洗脫,達到分離、純化和富集目標分析物的目的。固相萃取法目前已經被眾多研究者用于食用油中的多環芳烴的分離純化。固相萃取的大致操作流程如圖1所示。
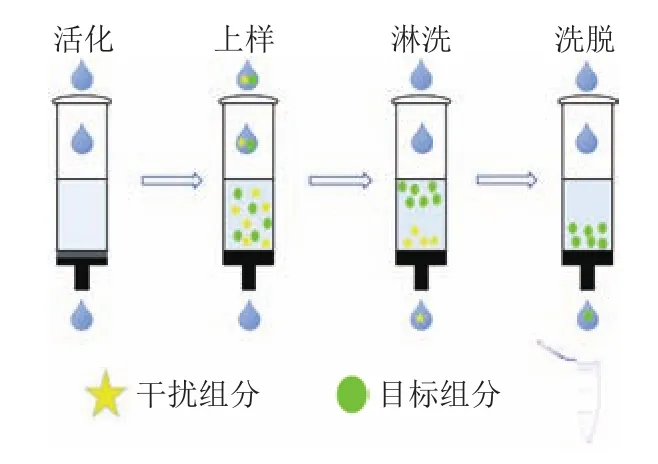
圖1 固相萃取的操作示意圖Fig.1 Schematic diagram of solid phase extraction
Shi Longkai等采用超聲輔助液液萃取,硅膠固相萃取柱純化,并通過GC-MS測定4種食用油脫臭餾出物中的16種多環芳烴,決定系數大于0.994,檢出限為0.06~0.13 μg/kg,定量限為0.18~0.42 μg/kg,回收率為84.8%~115.5%。Lee等采用液液萃取,硅膠固相萃取柱純化,并通過GC-MS測定芝麻油和紅辣椒油中的4種多環芳烴,決定系數大于0.996,檢出限為0.02~0.13 μg/kg,定量限為0.06~0.44 μg/kg,回收率為73.5%~112.6%,該方法時間較短,溶劑消耗少,背景基質干擾低,可以直接從油樣中分離多環芳烴。Stenerson等采用含有C與ZrO-SiO混合物和活性弗羅里硅土的雙層吸附固相萃取柱進行純化,結合HPLC-FLD檢測橄欖油中的16種多環芳烴,決定系數大于0.929,檢出限為0.2~1.0 μg/kg,定量限為0.65~3.40 μg/kg,回收率為79%~127%。Ju等采用液液萃取,經超聲、離心,運用EZ-POP NP雙層固相萃取柱和氨基固相萃取柱純化,充分去除了共萃取雜質,并結合ID-GC-高分辨率質譜(high resolution mass spectrometry,HRMS)檢測橄欖油中的4種多環芳烴,檢出限為0.08~0.10 μg/kg,定量限為0.10~0.28 μg/kg,回收率為97.5%~102%,結果可靠。
固相萃取法具有快速、準確、高效、有機溶劑用量少、選擇性高、可自動化等優點,不足之處是不能重復使用,成本較高。目前,許多學者開發了很多新型的固相吸附劑材料,提高了固相萃取的萃取效率,使固相萃取的應用更加廣泛,但更高效、更適用于油脂基質的高選擇性固相吸附劑材料有待進一步研究。
1.2.3 固相微萃取
固相微萃取是在固相萃取基礎上發展起來的一種新的前處理技術。固相微萃取是基于“相似相溶”的原理,利用待測組分在樣品溶液或者頂空氣體與萃取纖維涂層之間的分配平衡過程,達到萃取、富集的目的。根據測定的方式,固相微萃取技術可以分為3種:直接浸入式固相微萃取(direct extraction solid phase microextraction,DI-SPME)、頂空固相微萃取(headspace solid phase micro-extraction,HS-SPME)和中空纖維膜保護固相微萃取(hollow fiber membrane protected solid phase micro-extraction,HF-SPME)。以上3種固相微萃取方式的操作示意圖如圖2所示。
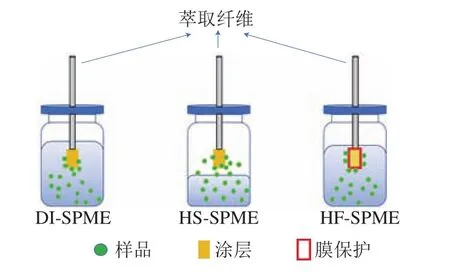
圖2 3種固相微萃取方式操作示意圖Fig.2 Schematic diagrams of three kinds of solid phase microextraction
Purcaro等采用DI-SPME技術,結合二維氣相色譜-飛行時間質譜法,測定橄欖油、橄欖果渣油、葵花籽油以及植物油中的16種多環芳烴,決定系數大于0.957,檢出限為0.2~1.4 μg/kg,定量限為0.4~4.6 μg/kg。此外,Purcaro等采用DI-SPME技術,結合GC-MS,測定橄欖油、橄欖果渣油、葡萄籽油以及4種谷物油中的10種多環芳烴,決定系數大于0.982,定量限為0.17~0.70 μg/kg,該方法減少了有機試劑消耗和甘油三酯的干擾,同時簡化了實驗操作。Chopra等通過HS-SPME技術結合GC-MS,測定魚油中的12種多環芳烴,方法的決定系數大于0.990,檢出限為1~7 μg/kg,定量限為3~21 μg/kg,回收率為80%~95%,該方法結果準確性較高,但在脂質復雜基質中平衡時間較慢。
固相微萃取靈敏度高,選擇性好,與固相萃取相比,有機試劑的用量更少,操作更為簡單,萃取富集的時間更短,回收率高,可實現固相萃取微型化,但是該方法的吸附容量有限,而且萃取頭一般為石英纖維,易損壞,限制了其使用壽命,其較少應用于油脂中多環芳烴的前處理。因此,開發更高效、更適用于脂質基質的萃取涂層材料和研發新的固相微萃取裝置解決萃取纖維頭的使用壽命及成本問題是目前該方法的兩個研究方向。
1.2.4 分散固相萃取
分散固相萃取法是在固相萃取基礎上發展起來的一種新型前處理技術。分散固相萃取的吸附原理與固相萃取相似,區別是在分散固相萃取中,吸附劑不再以萃取柱形式存在,而是直接分散在樣品萃取液中,通過振動、攪拌、超聲等方式,使吸附劑與目標組分相互作用,在達到吸附平衡后,通過離心過濾使已吸附目標組分的吸附劑與樣品基質分離,取上清液進行分析。固相萃取的操作如圖3所示。
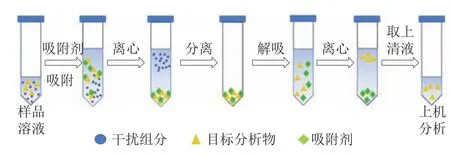
圖3 分散固相萃取的操作示意圖Fig.3 Schematic diagram of dispersive solid phase extraction
分散固相萃取最開始主要用于農藥殘留的提取,近些年在多環芳烴的檢測中也有應用。Zacs等以多壁碳納米管作為分散固相萃取吸附劑,對食用油樣品進行預處理,并結合GC-MS,測定食用油樣品中的4種多環芳烴,方法的決定系數大于0.983 3,檢出限為0.06~0.21 μg/kg,定量限為0.19~0.71 μg/kg,回收率為96%~107%。Lv Zhiyang等以多孔金屬有機骨架MIL-101(Cr)作為分散固相萃取吸附劑,對食用油樣品進行處理,并結合HPLC-FLD,測定花生油、大豆油、菜籽油、辣椒油中苯并[]芘,該方法減少了有機溶劑的消耗,簡化了操作,決定系數大于0.995 8,檢出限為0.19 μg/L,定量限為0.56 μg/L,回收率為79.6%~117.1%。
分散固相萃取法簡單快速、取樣量少、溶劑用量小、成本較低、易于操作、選擇性好,但是吸附容量有限,商業化和自動化程度較低。因此,需開發更高效、吸附容量更大、更適用于油脂基質的吸附劑材料,降低吸附劑材料的制備成本,研發分散固相萃取聯用技術及裝置,擴大其應用領域。
1.2.5 磁性固相萃取
磁性固相萃取是在分散固相萃取基礎上,以磁性材料或可磁化材料為吸附劑的一種前處理技術。磁性固相萃取的原理是利用磁性或可磁性化吸附劑富集目標物,在外加磁場的作用下達到分離、純化的目的。磁性固相萃取常用的材料有磁性碳基材料、離子液體、金屬有機骨架、分子印跡聚合物等。磁性固相萃取的操作如圖4所示。
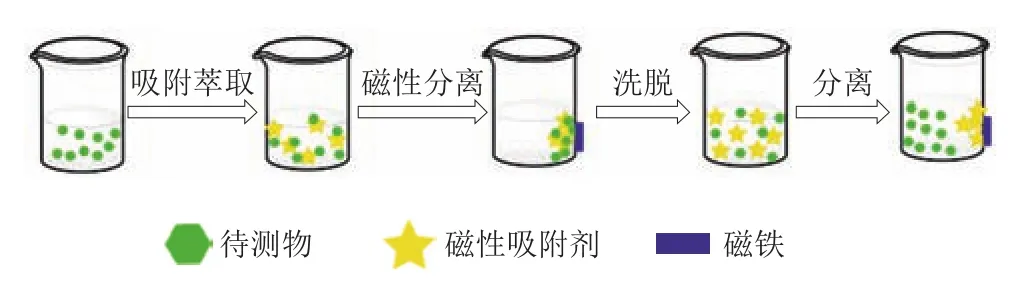
圖4 磁性固相萃取的操作示意圖Fig.4 Schematic diagram of magnetic solid phase extraction
Wang Qing等以磁性多壁碳納米管-十八烷基磷酸修飾氧化鋯為磁性吸附劑,采用磁性固相萃取法結合HPLC-DAD,測定花生油、大豆油、葵花籽油中的6種多環芳烴,該方法高效、低成本、富集能力強、靈敏度高、可重復使用,決定系數為0.993 7~0.999 4,檢出限為0.06~0.55 μg/kg,定量限為0.20~1.83 μg/kg,回收率為93.5%~113.2%。Zheng Haobo等以磁性氮化碳納米薄片為磁性吸附劑,采用磁性固相萃取法結合GC-MS,測定大豆油、玉米油、茶油、葵花籽油中的8種多環芳烴,該方法的相關系數為0.996 7~0.999 8,檢出限為0.1~0.3 μg/kg,定量限為0.4~0.9 μg/kg,其中在大豆油中回收率為91.0%~124.1%,其他3種食用油中回收率為79.1%~107.9%,該方法制備簡單、重現性好、耗時短,只需要10 min。Ji Wenhua等以植酸穩定的氧化鐵-石墨烯為磁性吸附劑,采用磁性固相萃取法,結合HPLC-DAD-UV-Vis,測定花生油、玉米油、大豆油和橄欖油中的8種多環芳烴,該方法的決定系數為0.998 9~0.999 9,檢出限為0.06~0.15 μg/kg,定量限為0.2~0.5 μg/kg,回收率為85.6%~102.3%。Zhang Yun等以三維離子液體功能化磁性氧化石墨烯納米復合材料為磁性吸附劑,采用磁性固相萃取法,結合GC-MS,測定花生油、大豆油、菜籽油、葵花籽油中的16種多環芳烴,該方法的決定系數大于0.999 2,檢出限為0.05~0.30 μg/kg,定量限為0.17~1.00 μg/kg,該方法溶劑消耗少,可重復使用,易與樣品溶液分離。
磁性固相萃取方法快速簡單、綠色友好,極大地簡化了前處理步驟,萃取速度快,便于回收,可重復使用,但尚未實現批量生產和自動化。今后的研究熱點是開發穩定性好、選擇性高、吸附容量大、富集能力強、萃取液使用量少、成本低的磁性吸附劑材料,推動其在食用油中多環芳烴檢測方面的快速化、商業化和自動化發展。
1.2.6 分子印跡萃取
分子印跡萃取是一種高效的前處理方法,其原理是通過合成分子印跡聚合物,以其為吸附劑,利用分子印跡聚合物的構效預定性、特異識別性和廣泛適用性,對目標組分進行吸附結合,達到分離純化的目的。分子印跡固相萃取的操作示意圖如圖5所示。
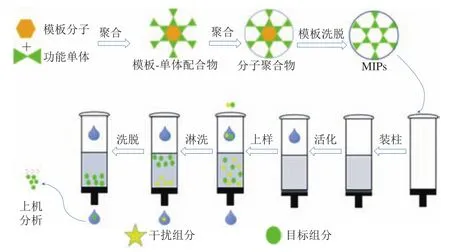
圖5 分子印跡固相萃取的操作示意圖Fig.5 Schematic diagram of molecularly imprinted solid phase extraction
Zhou Hua等采用分子印跡固相萃取柱和多環芳烴專用固相萃取柱聯用,分離純化食用油中的24種多環芳烴,并通過GC-MS進行檢測,該方法的相關系數大于0.999,檢出限為0.1~1.0 μg/kg,回收率為86%~116%。Sun Ying等采用分子印跡多環芳烴專用固相萃取柱和凝膠滲透色譜法分離純化脂質樣品中的16種多環芳烴,并用氧化鋁固相萃取柱進一步純化,結合GC-MS檢測,結果顯示分子印跡萃取法與凝膠滲透色譜法測定的回收率一樣高,且分子印跡萃取法的去除基質干擾效果更好,更有利于后續的GC-MS檢測。Xu Ting等采用分子印跡多環芳烴專用固相萃取柱和活性碳固相萃取柱聯用,分離純化食用油中的24種多環芳烴,并進行GC-MS檢測,該方法選擇性、靈敏度高,決定系數大于0.998,檢出限為0.03~0.60 μg/kg,定量限為0.13~2.00 μg/kg,回收率為56.8%~117.7%。Drabova等比較了凝膠滲透色譜、凝膠滲透色譜與硅膠固相萃取柱聯用、多環芳烴分子印跡固相萃取專用柱3種方法純化7種植物油中的16種多環芳烴的效果,其中多環芳烴分子印跡固相萃取專用柱的效果最好,定量限為0.1~0.3 μg/kg,回收率為70%~99%。
分子印跡技術能夠對目標組分進行特異性識別,制備簡單、溶劑消耗少、穩定性好、回收率高、檢出限低,但在實際應用中仍存在幾個問題:1)目前分子印跡技術多用于剛性小分子,且用于制備分子聚合物的一些模板分子和商品化的分子印跡固相萃取柱較貴,導致成本較高,未來可利用計算機模擬分子印跡聚合物的合成路線,節約制備成本,縮短制備時間,促進其商業化;2)分子印跡技術目前自動化程度較低,需加強分子印跡技術與檢測技術的聯用以促進其自動化。
1.3 分離、純化相結合方法
1.3.1 凝膠滲透色譜
凝膠滲透色譜又稱體積排阻色譜,是一種按分子尺寸大小的順序進行分離的一種色譜分析方法,可根據樣品中各組分分子質量的不同將其分離。由于油脂中主要成分甘油三酯的分子質量大于多環芳烴的分子質量,因此可用凝膠滲透色譜方法將多環芳烴從甘油三酯等基質中分離出來。
Wang等采用凝膠滲透色譜凈化,結合UHPLCDAD-FLD測定食用油中14種多環芳烴,該方法的決定系數大于0.999,檢出限為2.5~10.0 μg/kg,定量限為5~150 μg/kg,回收率為73%~110%。Zhu Huaming等通過環己烷-乙酸乙酯(體積比1∶1)混合溶液萃取,凝膠滲透色譜凈化,結合HPLC-FLD測定茶油中15種多環芳烴,該方法的相關系數大于0.991,檢出限為0.10~0.20 μg/kg,定量限為0.33~0.67 μg/kg,回收率為79.3%~97.3%。Wang Jianhua等以乙腈為溶劑,經過超聲提取、自制凝膠滲透色譜柱凈化,結合ID-GC-MS測定植物油中的16種多環芳烴,該方法的定量限為0.3~0.6 μg/kg,回收率為81%~96%,其自制的短窄凝膠滲透色譜柱可以顯著減少溶劑和凝膠滲透色譜樹脂的用量。Fromberg等采用凝膠滲透色譜和固相萃取技術相結合,并通過GC-MS測定食用油中24種多環芳烴,檢出限為0.2~1.5 μg/kg,定量限為0.3~3.0 μg/kg,回收率為59%~120%。Ballesteros等采用液液萃取和凝膠滲透色譜凈化相結合,結合GC-MS,同時測定初榨橄欖油、精制橄欖油和橄欖果榨油中的農藥殘留和4種多環芳烴,該方法凈化效果好,其中多環芳烴測定的決定系數大于0.992,檢出限為0.05~0.07 μg/kg,定量限為0.10~0.20 μg/kg,回收率為84%~110%。
凝膠滲透色譜將分離、純化相結合,方法簡單快速、易于操作、可自動化、回收率高、重復性好、穩定性好,被廣泛應用于油脂中多環芳烴的分離純化,但設備成本高、有機溶劑消耗大、耗時長。如何減少溶劑消耗、加快分析速率是目前凝膠滲透色譜亟待解決的問題。
1.3.2 QuEChERS前處理技術
Anastassiades等首次提出了QuEChERS方法,QuEChERS基于液液萃取和分散固相萃取的原理,利用吸附劑來富集目標物,提取目標組分,達到除雜凈化的目的,是一種快速、簡單、廉價、高效、安全的樣品前處理技術。QuEChERS前處理技術的操作如圖6所示。
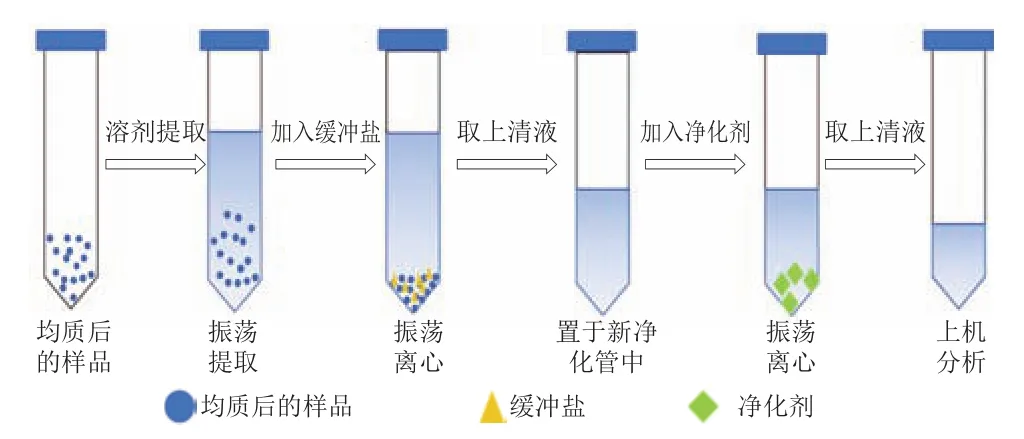
圖6 QuEChERS的操作示意圖Fig.6 Schematic diagram of QuEChERS
QuEChERS技術最開始用于農藥殘留的檢測,只能處理果蔬等基質較為簡單的樣品,而對于脂質含量較高的樣品凈化效果不太理想。處理油脂樣品的關鍵是選擇合適的溶劑和吸附劑,減少油中共提物的干擾,在QuEChERS方法中,常用的吸附劑有C、-丙基乙二胺(primary secondary amine,PSA)、石墨化炭黑(graphitized carbon black,GCB)、增強型基質去除(enhanced matrix removal,EMR)。
Sun Yaqing等采用EMR為吸附劑填料,利用乙腈-丙酮(體積比為3∶2)的混合溶液萃取,結合GC-三重四極桿(triple quadrupole,QqQ)-MS同時測定廢棄煎炸油中的16種多環芳烴,該方法的決定系數大于0.995,檢出限為0.06~0.13 μg/L,定量限為0.20~0.43 μg/L,回收率為66.72%~112.87%,提高了重質多環芳烴的回收率,減少了溶劑使用量,并實現了快速檢測。朱捷等采用以PSA和C為吸附劑填料的QuEChERS方法,選擇乙腈作為萃取試劑,結合GC-QqQ-MS同時檢測枸杞籽油中16種多環芳烴,結果表明決定系數大于0.998,檢出限為0.2~3.5 μg/kg,定量限為0.7~11.5 μg/kg,回收率為60.04%~119.00%,靈敏度高、結果較好。胡國紳等比較了以EMR吸附劑作為填料的QuEChERS法和凝膠滲透色譜法的分離效果,結合GC-MS測定植物油中的16種多環芳烴,與凝膠滲透色譜的凈化效果相比較,QuEChERS測定結果的準確性和精密度較高,溶劑使用量較少,且決定系數大于0.995,定量限為0.3~0.8 μg/kg,回收率為88.9%~112.3%。
與其他方法相比,QuEChERS方法快速、簡便、可靠、溶劑消耗少、樣品制備相對便宜、回收率較為理想,是一種高通量的綠色前處理方法,但吸附劑的吸附容量有限。因此開發更高效的吸附劑材料、提高其吸附容量和重復使用次數仍是目前有待解決的問題,對進一步提高QuEChERS方法在快速分離富集食用油中多環芳烴方面的應用有重要意義。
除此之外,其他一些前處理方法,如加速溶劑萃取法、薄層色譜法、供體受體復合色譜、泡騰輔助微萃取、分散液液微萃取等也開始應用于食用油多環芳烴的分析中。
上述食用油中多環芳烴檢測的樣品前處理方法都具有較好的萃取效率和回收率,但是各前處理方法的萃取效果還是存在差異。表1總結了不同分離、純化食用油中多環芳烴方法的優缺點。
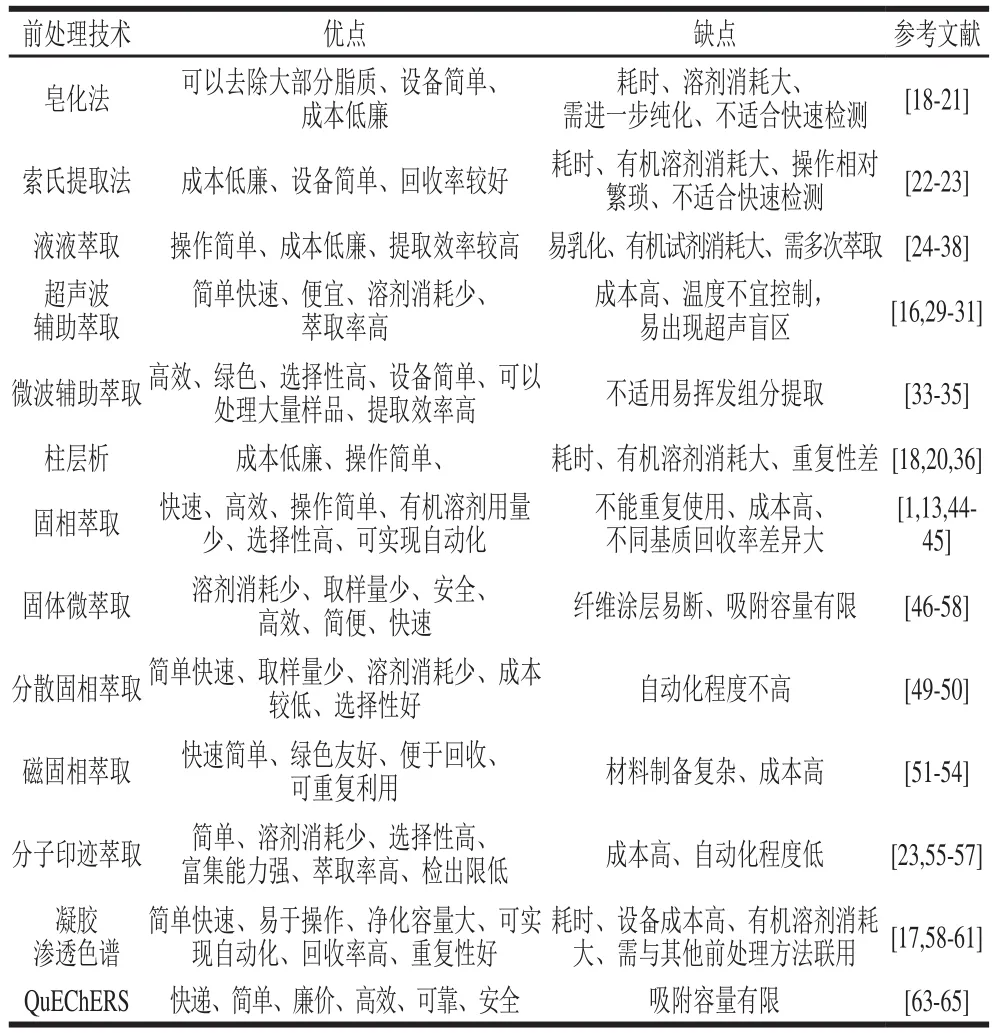
表1 不同前處理方法提取多環芳烴的優缺點對比Table 1 Comparison of advantages and disadvantages of different pretreatment methods for PAHs extraction
2 結 語
本文重點綜述了近年來食用油中多環芳烴檢測過程中的樣品前處理技術。傳統的前處理方法如皂化法、索氏提取法、液液萃取法、柱層析法成本低廉,不需要復雜的設備,但是處理時間長、有機溶劑使用量大、回收率低、穩定性差,越來越難滿足目前檢測的需求。新型的前處理技術如固相微萃取技術、分散固相萃取技術、分子印跡萃取技術、磁性固相萃取技術快速簡單、靈敏度高、溶劑使用量少、回收率高,但是吸附容量有限,且自動化和微型化程度低。雖然新型的萃取技術已被應用于從食用油中分離純化多環芳烴,但更高效、快速、綠色、高通量、能夠實現現場便攜式檢測的前處理技術仍有待開發。
建立方便、可靠、靈敏、快速、綠色無污染的前處理方法,實現設備自動化、小型化和便攜化,始終是食用油中多環芳烴的檢測前處理方法的研究趨勢。未來食用油中多環芳烴檢測前處理方法的研究可在以下幾個方面實現突破:采用多種前處理方法聯用,提高分離純化的效率;開發新型的、吸附量高、更適用于脂質基質的吸附劑材料,提高靈敏度,促進商業化;開發新的前處理方法,簡化前處理過程,實現自動化、小型化、便攜化,促進其在現場快速檢測中的應用。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
