肯尼迪病5例臨床特征分析并文獻復習
2022-07-06 07:27:48錢曉鋒張堯時伊初祁云潔孫歡季鄭軍李則衡葛巍
東南大學學報(醫學版) 2022年3期
錢曉鋒,張堯,時伊初,祁云潔,孫歡,季鄭軍,李則衡,葛巍
(1.徐州醫科大學 研究生院,江蘇 徐州 221004; 2.徐州醫科大學附屬醫院 心內科,江蘇 徐州 221002;3.南京市第一醫院,江蘇 南京 210012; 4.徐州醫科大學附屬醫院 神經內科,江蘇 徐州 221002)
肯尼迪病(Kennedy’s disease,KD)又稱為脊髓延髓肌萎縮癥(spinal and bulbar muscular atrophy,SBMA),是一種以脊髓和延髓支配肌肉無力、萎縮為主要表現,可伴有全身多系統受累的遺傳性神經肌肉疾病[1]。該病最早由美國Kennedy等[2]于1968年首次描述并報道。1991年Spada等[3]發現,此病的分子遺傳學病因是X染色體上雄激素受體基因(androgen receptor,AR)第1個外顯子中的三核苷酸(CAG)序列異常擴增[3]。該病發病率較低,約為1/40 000[4],早期癥狀、體征缺乏特異性,尤其容易被誤診為其他運動神經元病。約有2%的肌萎縮側索硬化(ALS)患者實際上患有KD[5]。目前國內關于KD的報道較少,本研究選取近年來徐州醫科大學附屬醫院(下稱“本院”)收治的肯尼迪病患者,結合國內外相關文獻總結其臨床特征,以增強臨床醫師對該病的認識。
1 對象與方法
1.1 研究對象
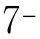
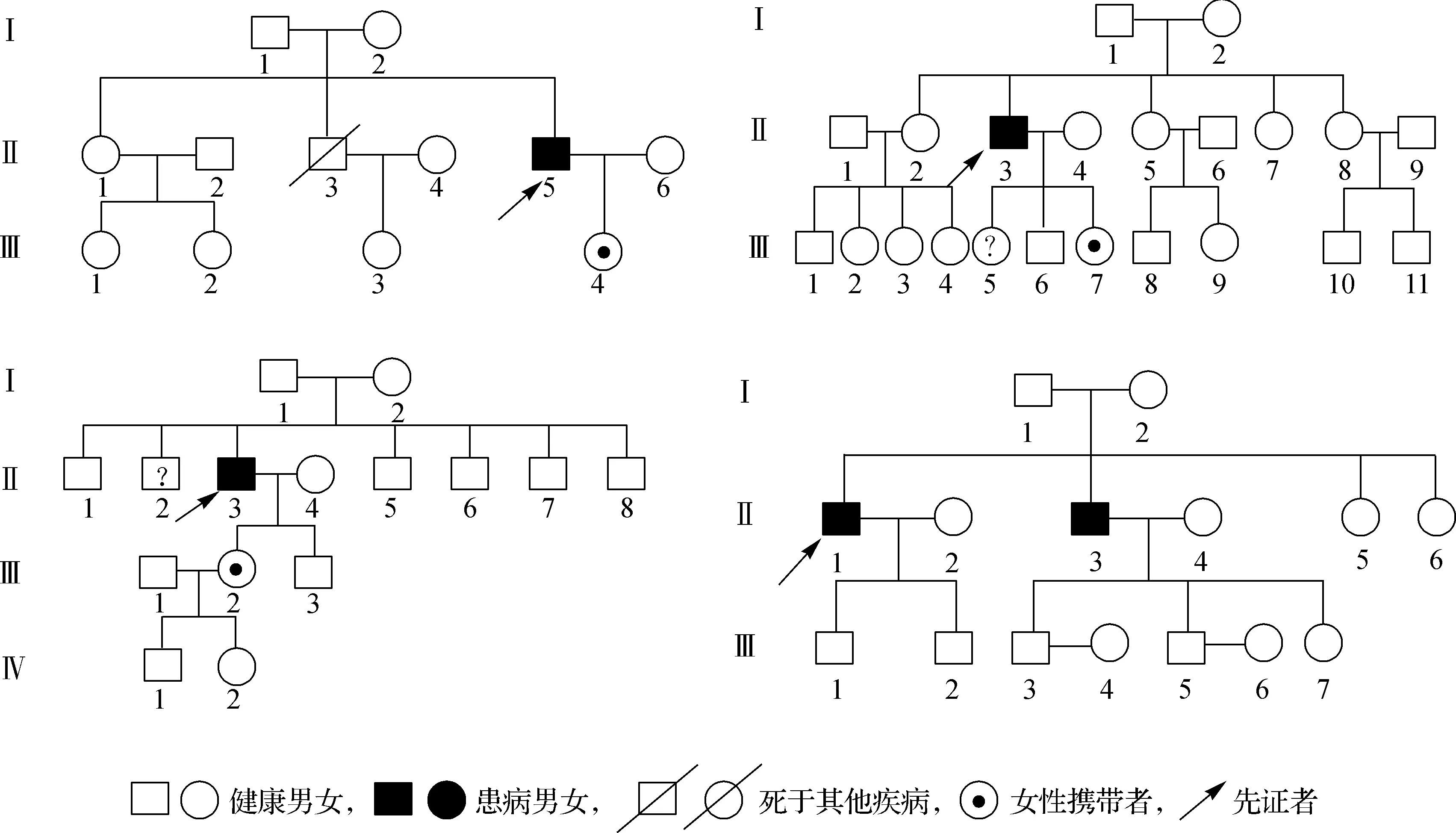
圖1 4個KD家系遺傳譜系圖
1.1.1 患者1 男,52歲。患者1995年出現雙上肢不自主抖動,不影響日常生活,未予重視;2006年出現雙下肢無力,易疲勞,伴全身多處“肉跳”,緩慢進行性加重;2017年出現活動后憋喘、爬樓費力,最多步行2 000 m;2018年步行距離下降,無法獨自上樓,于康旭醫學檢驗所行AR基因動態突變檢測示CAG重復次數為45次;2019年開始使用輪椅;現進展至四肢無力,表現為行走不穩、提重物困難,最多步行50 m,偶有飲水嗆咳。近4年來體質量增加23.5 kg。體檢:體質量指數(BMI)為32.35 kg·m-2,肥胖貌,乳房增大。構音障礙,舌肌萎縮伴纖顫,可見口周肌束震顫。軀干及四肢肌肉萎縮,近端為主,右上肢及雙下肢近端肌力4級,遠端4+級,左上肢肌力4級;雙手姿勢性震顫;上肢腱反射(+),下肢腱反射(-),雙膝關節震動覺消失,雙側病理征(-)。
1.1.2 患者2 男,60歲。2001年出現雙上肢震顫,不影響日常生活;2006年出現雙下肢乏力,表現為活動后腿沉,緩慢進行性加重;2017年癥狀明顯加重前來就診,就診時發現患者言語不清、乳房增大;現雙下肢無力癥狀較前有所改善,蹲下起立費力,可獨自上一層樓,期間未予特殊治療。體檢:BMI為26.95 kg·m-2,腹型肥胖,雙乳房增大,右側明顯。構音障礙,可見舌肌萎縮、舌肌纖顫及口周肌束震顫。雙上肢近端肌肉萎縮,四肢肌力近端4+級,遠端5級,雙上肢可見震顫,四肢腱反射(-),雙側病理征(-)。
1.1.3 患者3 男,67歲。2009年出現下肢無力,上樓較同齡人慢,活動后易疲勞;2014年出現言語不清、雙上肢無力,伴全身“肉跳”;2016年出現上坡費力,同年發現肌肉萎縮;2017年因四肢無力加重伴雙手震顫來本院就診;2019年開始使用拐杖;現出現咀嚼無力,最多步行約500 m。體檢:BMI為19.05 kg·m-2,體型消瘦,左乳房增大。輕度構音障礙,舌肌萎縮、纖顫,頸部可見肌束震顫。軀干肌及四肢肌肉萎縮,上肢近段及第一骨間肌萎縮顯著。四肢肌力近端4級,遠端4+級;四肢腱反射未引出,雙側病理征(-)。
1.1.4 患者4 男,63歲。1996年出現言語不清,說話帶鼻音,未予重視;1998年出現雙下肢無力,右下肢較重,按“腰椎間盤突出”治療無好轉;2004年出現雙手姿勢性震顫,主要表現為持筷不穩,按照“帕金森病”治療,癥狀無改善;2008年自覺行走不穩,易疲勞,尚不影響日常生活;2011年出現四肢無力,表現為四肢抬舉費力,蹲下起立困難,爬樓梯須借助扶手,伴右側肢體麻木;2016年開始借助拐杖活動,偶有飲水嗆咳及吞咽困難;2017年因肢體無力癥狀加重來本院就診;現可獨立步行10 m多,易跌倒,生活尚自理。體檢:BMI為28.21 kg·m-2,乳房增大,左側明顯,舌肌萎縮,舌肌纖顫。構音障礙,口周、面部及四肢均可見肌束震顫。四肢近端肌肉輕度萎縮,四肢肌力近端4級,遠端4+級,雙手姿勢性震顫,雙上肢腱反射(+),雙下肢腱反射(-),雙側病理征(-)。
1.1.5 患者5 男,57歲。1996年出現輕度聲音嘶啞、手部震顫及下肢無力,易疲勞,自覺步行速度較常人稍慢;2007年下肢無力較前加重,逐漸累及上肢,表現為爬樓及持重物困難;現說話略帶鼻音,日常活動須借助拐杖或輪椅。體檢:BMI為22.76 kg·m-2,雙側乳房增大,右側明顯。舌肌萎縮、纖顫,構音障礙,口周及四肢可見肌束震顫。四肢肌肉萎縮,近端為主,雙手姿勢性震顫,右上肢及雙下肢肌力4- 4- 4+- 4+級,左上肢肌力4+- 4+- 5- 5級,雙上肢腱反射(+),雙下肢腱反射(-),四肢病理征(-)。
1.2 方法
本研究將肢體無力或球部癥狀(如言語不清)出現時的年齡作為患者的發病年齡。完善血生化(肝腎功能、肌酶譜、血脂)、糖化血紅蛋白、性激素五項、肌電圖等檢查,除患者5以外其余患者均進行下肢肌肉磁共振檢查。另抽取患者及部分家屬外周血3 ml,EDTA抗凝,-20 ℃保存。從外周血白細胞中提取DNA,參考既往文獻設計引物(由上海生物工程有限公司合成),使用PCR法擴增目的基因片段,瓊脂糖凝膠電泳分析PCR產物片段數量及大小,對有意義的片段進一步行Sanger測序得到CAG重復數。所有受試者簽署知情同意書。
1.3 結果
患者均為男,起病年齡32~55歲,平均41.4歲,以肢體無力、肌肉萎縮、構音障礙、肉跳、手部震顫及乳房發育為主要臨床表現(圖2、3),1例伴有呼吸困難。所有患者肌酸激酶均有不同程度升高,4例患者肌酐低于正常值下限,3例患者出現血脂異常,1例患者出現肝轉氨酶異常,2例患者糖化血紅蛋白水平升高,4例患者出現性激素水平異常(表1、2)。大腿肌肉磁共振提示肌肉萎縮、脂肪浸潤。肌電圖檢查(表3)提示神經源性損害,運動神經及感覺神經均受累,2例患者高頻RNS波幅見遞減。Sanger測序顯示,CAG序列重重次數異常(圖4)。
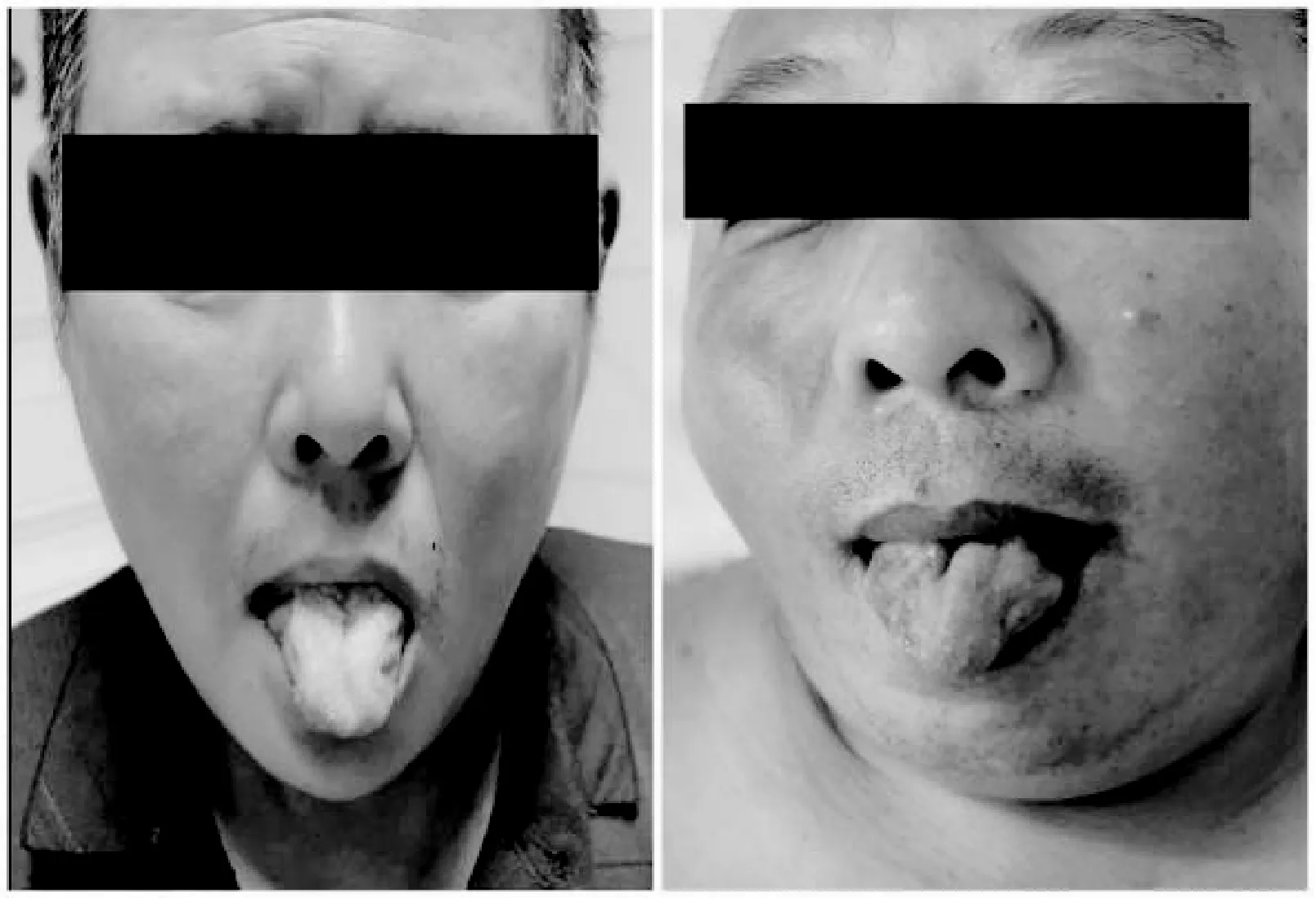
圖2 舌肌萎縮明顯
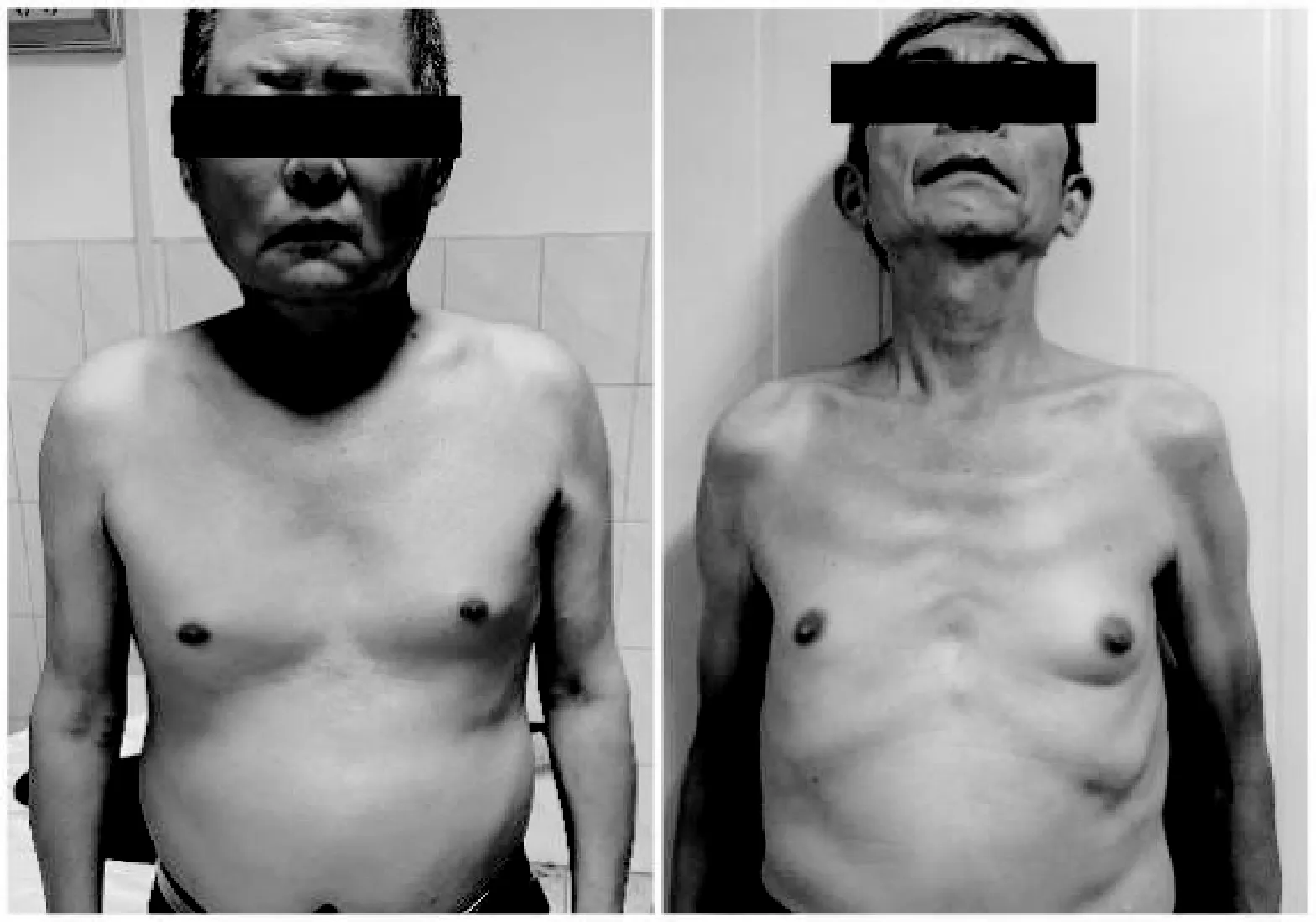
圖3 乳房發育
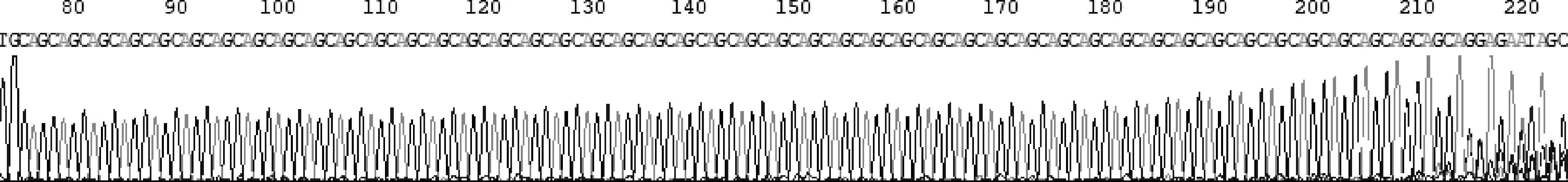
圖4 患者4的基因檢測
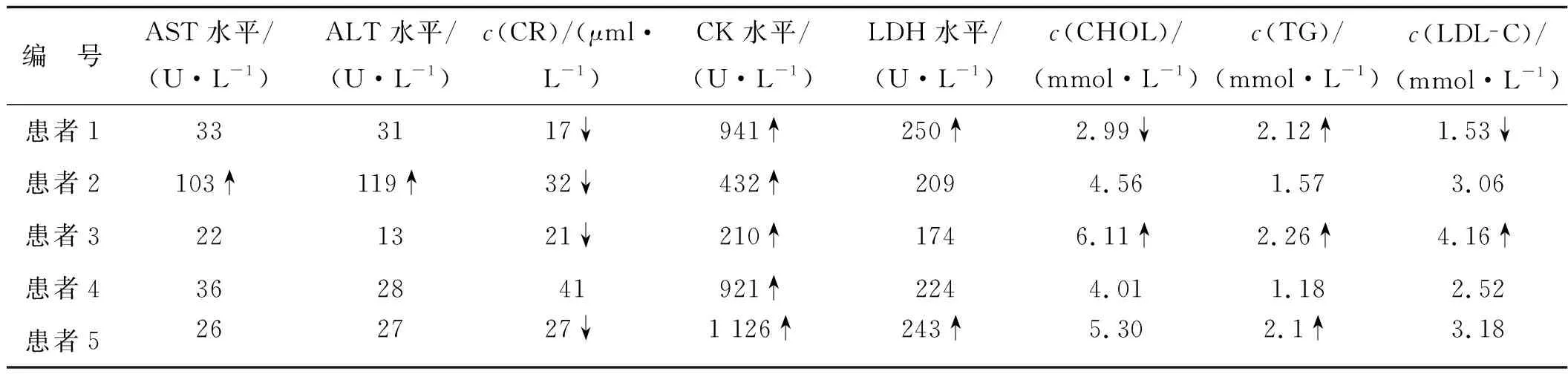
表1 KD患者的生化指標
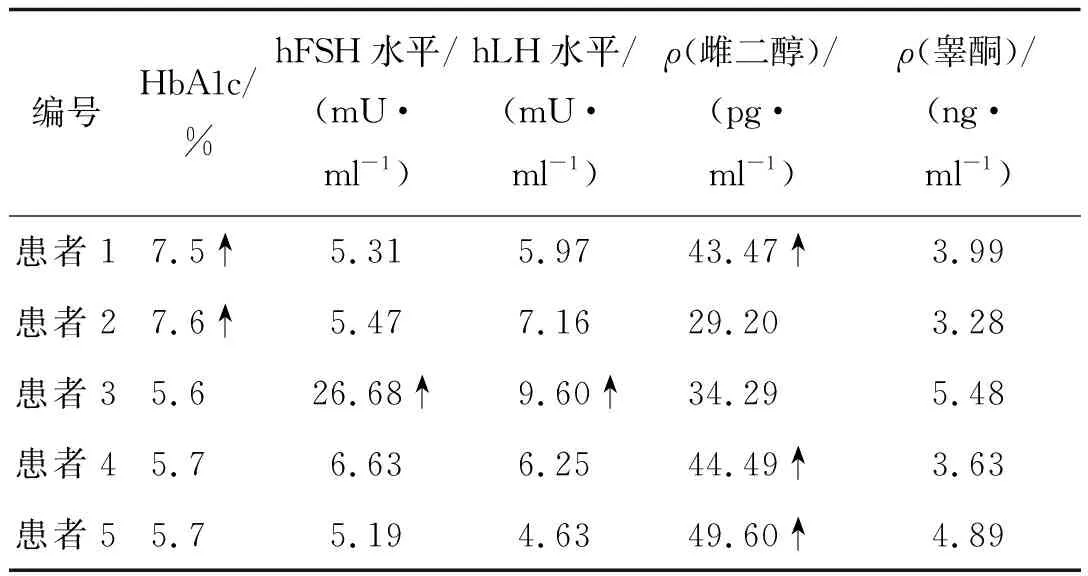
表2 患者的糖化血紅蛋白及性激素水平
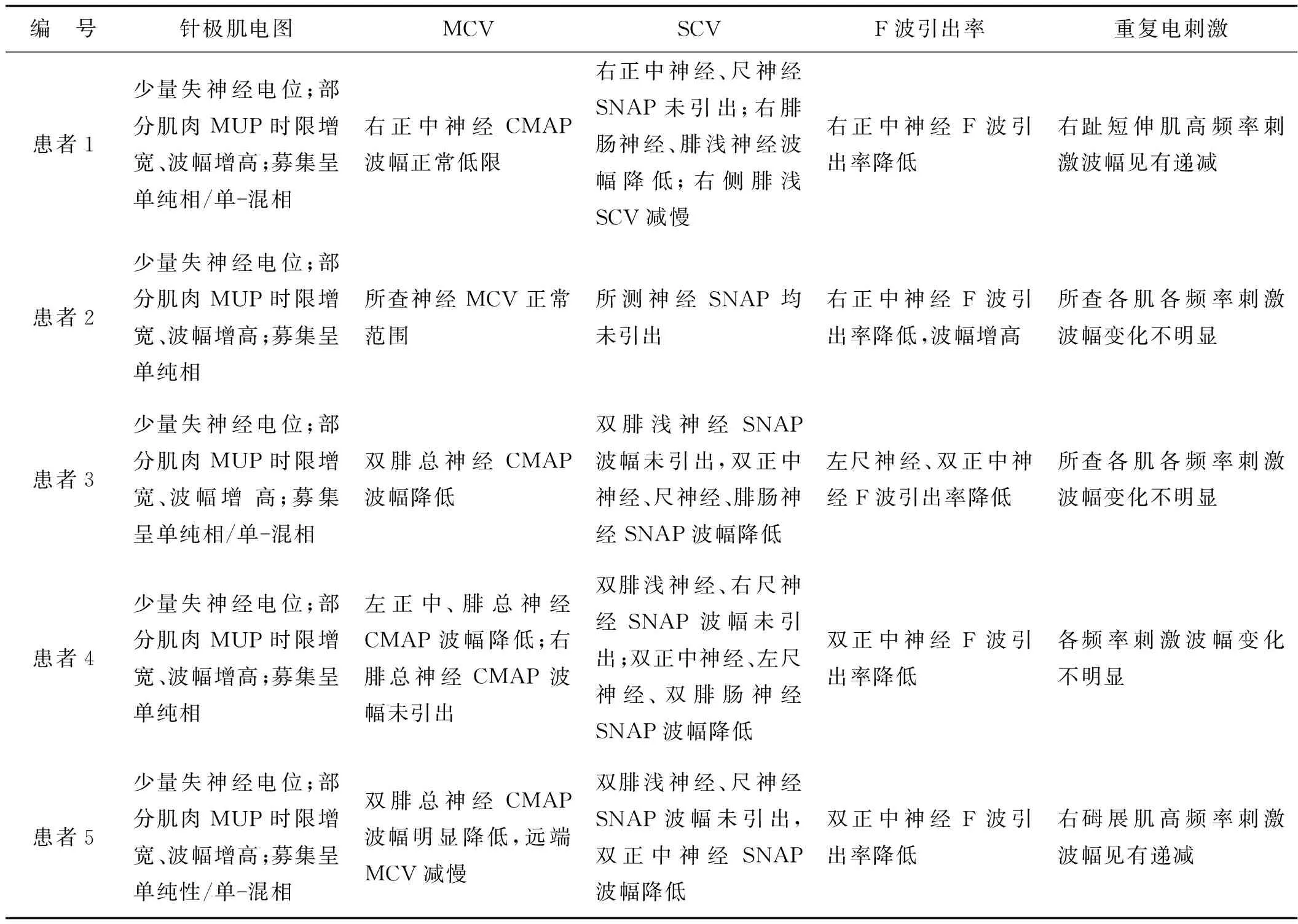
表3 患者的電生理表現
2 討 論
KD是一種微衛星(三核苷酸)擴張性疾病,與亨廷頓病(HD)、齒狀核紅核蒼白球路易體萎縮癥(DRPLA)以及脊髓小腦共濟失調(SCA)1、2、3、6、7和17型同屬于多聚谷氨酰胺(poly Q)類疾病[6]。該病主要累及下運動神經元,也有少數患者可出現腱反射活躍、病理征陽性等上運動神經元受損的表現[7]。患者的發病年齡差異較大,通常30~50歲,目前國外報道的KD起病年齡4~76歲[8]。肌無力是本病的核心臨床表現,出現在97%的KD患者中。約70%的KD患者運動障礙最初出現在下肢,伴有腱反射減弱或缺失[9],與其他運動神經元病相比(如肌萎縮側索硬化),疾病進展相對緩慢,肌肉力量每年大約下降2%[10]。由于球麻痹,KD患者存在窒息和吸入性肺炎的風險,但患者的預期壽命與正常人群并無顯著差異。本病由AR基因中CAG重復序列異常擴增所致,但確切致病機制尚不明確,仍在探索之中。脊髓和腦干運動核團中的神經元丟失以及核內包涵體形成并聚集是其主要的組織病理學表現。正常人群中CAG序列重復數一般為14~32次,而KD患者多≥38次。2014年歐洲神經科學聯合會指南則將CAG重復數≥35作為脊髓延髓肌萎縮癥的診斷依據,而CAG重復數為35~37的人群應根據有無發病癥狀、有無陽性家族史以及有無可疑的輔助檢查結果定期進行評估。雖然目前基因檢測技術已經逐漸得到推廣與普及,但由于對于本病的認識普遍不足,診斷仍較困難。
本研究中患者的起病年齡與起病方式與既往報道一致。值得注意的是,其中1例患者雖為雙下肢同時起病,但發病時兩側肌無力程度不對稱,且患者體型肥胖,肉眼觀察肌肉萎縮不明顯,早期被誤診為“腰椎間盤突出癥”,臨床診療過程中須警惕以類似癥狀起病的患者。球部受累也是KD的主要表現之一,5例患者均存在輕度構音障礙,與之形成鮮明對比的是,患者的舌肌萎縮十分顯著,舌中溝可見,甚至呈“花束狀”(圖2)。輕度的構音障礙或吞咽困難和相當明顯的舌肌萎縮是KD具有標志性意義的體征[11]。此外,部分患者可能由于口周肌束震顫而出現下巴的不自主抖動,也稱為“顫抖下巴”,在其他神經系統疾病中十分少見。本研究中,患者均被觀察到不同程度的乳房增大(圖3),考慮系對雄激素不敏感所致。雄激素不敏感跡象也是診斷KD的重要線索。高達73%~78%的KD患者存在男性乳房女性化,是KD最常見的神經系統外癥狀;約60%的KD患者伴有睪丸萎縮或少精子癥,勃起功能障礙和性欲減退在KD患者中也很十分常見[12]。

綜上所述,KD并非是簡單的神經系統遺傳性疾病,而是一種多系統受累疾病。KD發病率低,且癥狀、體征多樣,給其診斷帶來了挑戰。在診療過程中,對于具有類似表現的患者均應想到KD的可能性,及早完善基因檢測明確診斷。雖然目前KD尚缺乏特異性的治療方法,但早期確診并提供必要的遺傳咨詢以減少患兒的出生,對減輕家庭及社會負擔意義重大。
