基于化學計量學的多花黃精多糖部分酸水解產物PMP-HPLC 指紋圖譜構建
2022-07-08 13:54:46周忠瑜杜澤飛蒲婷婷楊麗英晏仁義段寶忠
食品工業科技 2022年13期
周忠瑜,杜澤飛,蒲婷婷,楊麗英,晏仁義,段寶忠,
(1.大理大學藥學院,云南大理 671000;2.云南省農業科學院藥用植物研究所,云南昆明 650205;3.湖北漢江大健康產業有限公司,湖北十堰 442521)
多花黃精為百合科黃精屬植物Polygonatum cyrtonemaHua.,又名姜狀黃精,為2020 年版《中華人民共和國藥典》黃精藥材的基原植物之一,是目前市場上黃精的主流栽培品種,具有補氣養陰,健脾,潤肺,益腎之功能[1],多花黃精為重要的藥食同源資源,由于其良好的功效,近年來被廣泛用于食品和保健品開發[2]。現代研究表明,多花黃精化學成分主要包括多糖,甾體皂苷、黃酮等成分[3?5],其中多糖是多花黃精中的質量標志物,也是中國藥典黃精的質量控制指標[1],具有抗衰老、降血糖、降血脂、抗菌等藥理作用[6?8]。據對保健食品原料的調查發現,黃精在中藥類原料的使用頻率排名靠前[9],目前市場上存在大量的黃精多糖產品如黃精膏、黃精飲料等產品,因此判定多花黃精產品的質量好壞和原料真偽極為重要。現有方法一般采用苯酚-硫酸法和蒽酮-硫酸法測定總多糖含量[10],用于控制多花黃精多糖產品質量,但此類方法選擇性較差,不能有效地鑒別出摻假品;因此,迫切需要制定有效可靠的評價標準,以便科學地評價多花黃精多糖的質量,對黃精多糖保健食品的應用開發具有深遠意義。
指紋圖譜是近年來用于中藥質量控制的一種有效、可行的方法,已廣泛用于中藥質量控制、藥效成分研究領域[11?13]。由于黃精多糖為非單一分子化合物,結構復雜,且不含有共軛結構,使用常規高效液相二極管陣列檢測法(High-performance liquid chromatography-diode array detection,HPLC-DAD)方法無法有效檢測。已有學者采用高效液相色譜-蒸發光散射法、高效陰離子交換色譜-脈沖安培法和電噴霧式檢測器(Charged aerosol detector,CAD)對植物多糖的單糖組成進行了研究[14?16],但蒸發光散射檢測的耐用性和穩定性欠佳[17?18],脈沖安培檢測的成本較高、穩定性差且對色譜條件要求較為嚴格[16],CAD 檢測儀器價格昂貴,使用受到一定的限制[19?21]。有關黃精法定基原植物的指紋圖譜研究方面,目前已有學者對滇黃精P.kingianumColl.Et Hemsl.和黃精P.sibiricumRed.多糖的指紋圖譜進行了研究[22?23];而對于多花黃精P.cyrtonema的指紋圖譜研究方面,僅見周寶珍,楊青等對多花黃精乙醇提取物的指紋圖譜研究[24?25],尚未見多花黃精中的多糖組分的指紋圖譜的報道。因此,建立能表征多花黃精中多糖指紋的方法具有重要意義。鑒于此,本文采用柱前衍生化-HPLC 對多花黃精藥材的指紋圖譜和單糖組成進行研究,并結合相似度分析(Similarity analysis,SA)、聚類分析(Hi-erarchical cluster analysis,HCA)和主成分分析(Prin-cipal component analysis,PCA)等化學計量學[26?28]手段,以期為多花黃精藥材的質量控制奠定科學基礎。
1 材料與方法
1.1 材料與儀器
黃精藥材 13 批樣品 購于安徽、四川、湖南等地,均為栽培品,經大理大學段寶忠教授鑒定為百合科植物多花黃精P.cyrtonemaHua.,新鮮根莖切片后于40 ℃烘箱烘干,樣品信息詳見表1,憑證標本保存于大理大學中藥標本館;葡萄糖(批號171106)、甘露糖(批號170921)、鹽酸氨基葡萄糖(批號171210)、半乳糖(批號171206)、L-鼠李糖(批號171024)、L-巖藻糖(批號170813)、葡萄糖醛酸(批號170730)、木糖(批號170912)、半乳糖醛酸(批號170903)、核糖(批號171103)、阿拉伯糖(批號171219) 以上單糖純度≥98%,購自上海融禾醫藥科技有限公司;1-苯基-3-甲基-5-吡唑啉酮(1-pheny-3-methyl-5-pyrazolone,PMP) 阿拉丁;色譜級乙腈(批號085884) Fisher Scientific;水為超純水,其他試劑為國產分析純。
表1 樣品信息Table 1 Information of samples
Agilent 1200 高效液相色譜儀 美國安捷倫科技有限公司;GH-252 電子天平 日本AND 公司;AL204 電子天平 梅特勒-托利多儀器(上海)有限公司;SB25-12D 超聲波清洗機 寧波新芝生物科技股份有限公司。
1.2 實驗方法
1.2.1 色譜條件 Agilent Zorbax SB-C18色譜柱(250 mm×4.6 mm,5 μm);流動相:乙腈(A)?0.025 mol·L?1磷酸鹽緩沖溶液(B)(pH7.5);梯度洗脫(0~10 min,15%~17% A;10~18.5 min,17%~22.5%A;18.5~20 min,22.5%~23.5% A;20~32 min,23.5%~30% A);流速:0.8 mL·min?1;柱溫:35 ℃;檢測波長:250 nm;進樣量:20 μL。
1.2.2 衍生化對照品溶液制備 精密稱取各單糖對照品適量,用蒸餾水配制成濃度約為0.5 mg·mL?1的單糖對照品溶液。精密吸取上述對照品溶液400 μL,置于5 mL 的安瓿瓶中,精密加入200 μL 0.6 mol·L?1NaOH 和0.5 mol·L?1PMP 溶液,置70 ℃條件下反應60 min。取出,放冷,精密加入200 μL 0.6 mol·L?1HCl 溶液,混勻。加入等體積的三氯甲烷,混勻,離心10 min(4000 r·min?1),去掉三氯甲烷層,重復多次至三氯甲烷層無色,即得衍生化對照品溶液。
1.2.3 供試品溶液制備
1.2.3.1 粗多糖制備 取多花黃精樣品于40 ℃烘箱干燥12 h 至恒重,粉碎,過80 目篩,取粉末5.0 g,精密稱定,根據預實驗得到最優工藝制備粗多糖,即加入100 mL 水于80 ℃下超聲提取,重復3 次,每次1 h,抽濾,洗凈濾渣,濃縮至10 mL,放入離心機離心20 min(4000 r·min?1)。取上清液轉移至分液漏斗,加入3 倍體積的石油醚進行萃取,直至石油醚層無色,靜置分層,取下層水相,調節pH 至6,加入體積分數為2%的木瓜蛋白酶溶液(80 萬U/g),水浴溫度60 ℃,酶解4 h,待反應完成,沸水浴滅酶10 min。取上清液采用Sevage 法(正丁醇:氯仿=1:5)脫去蛋白,直至無絮狀物生成,離心10 min(4000 r·min?1),再取上清液轉至燒杯中,精密緩慢的加入6 倍量的無水乙醇,快速攪拌,于4 ℃冰箱中放置12 h,離心,向沉淀中加10 mL 95%乙醇,洗滌2 次,再離心,加熱水使沉淀溶解,轉移至10 mL 量瓶中,室溫下放冷,后定容,再將溶液置于燒杯中冷凍干燥,最后得粗多糖粉末。
1.2.3.2 衍生化供試品溶液制備 精密稱取5 mg 粗多糖粉末,置于5 mL 的安瓿瓶中,精密加入2 mL 4 mol·L?1三氟乙酸(TFA)溶液,封口,置于110 ℃條件下7 h,進行水解,取出,室溫放冷,后水浴蒸干,向殘渣中加入甲醇1 mL,烘干,重復多次,直至三氟乙酸除盡。再加熱水適量,使沉淀溶解,轉移至1 mL容量瓶中,室溫放冷,定容搖勻,得供試品酸水解溶液。取酸水解溶液400 μL,按1.2.2 項下衍生化方法制備,得衍生化供試品溶液,每批樣品重復3 次。
1.2.4 總多糖含量測定 按課題組已發表文獻方法,對13 批多花黃精總多糖含量進行測定[10]。
1.3 數據處理
相似度分析采用《中藥色譜指紋圖譜相似度評價系統》(2.0 版),聚類分析采用SPSS 20.0 軟件進行,主成分分析采用SIMCA 13.0 軟件進行。
2 結果與分析
2.1 色譜條件的選擇
本研究比較了以下4 種色譜柱:Zorbax SB-C18(4.6 mm×250 mm,5 μm),Zorbax Extend-C18(4.6 mm×250 mm,5 μm),Zorbax SB-Aq-C18(4.6 mm×250 mm,5 μm),Zorbax XDB-C18(4.6 mm×250 mm,5 μm)。實驗結果表明,Agilent Zorbax SB-C18色譜柱(4.6 mm×250 mm,5 μm)出峰較多,各成分分離較好。同時,當流動相為純水時,部分色譜峰產生了拖尾情況,因此,在實驗中考察不同濃度磷酸鹽緩沖溶液(0.01、0.02、0.025 mol·L?1)對色譜峰分離效果的影響,結果顯示,當磷酸鹽緩沖溶液濃度為0.025 mol·L?1時,所得色譜峰峰形及分離度較好,故選擇0.025 mol·L?1的磷酸鹽緩沖溶液作為洗脫溶劑。
2.2 方法學考察
2.2.1 精密度試驗 取同一批供試品溶液(S2),按1.2.1 項下方法連續進樣6 次,測得各共有峰相對保留時間和相對峰面積的RSD 均小于2.10%,表明精密度良好。
2.2.2 重復性試驗 取同一批S2 樣品6 份,按1.2.3項下方法進行制備,在1.2.1 項色譜條件下進樣分析。測得結果顯示,各共有峰的相對保留時間與相對峰面積的RSD 均小于2.60%,表明重復性良好。
2.2.3 穩定性試驗 取同一批S2 供試品溶液,分別在0、4、8、12、18、24、48 h 進樣測定,測得結果顯示,各共有峰的相對保留時間與相對峰面積的RSD均小于2.70%,表明供試品溶液在48 h 內穩定。
2.3 指紋圖譜的構建
將13 批多花黃精樣品按1.2.3 方法制備供試品溶液,按1.2.1 項色譜條件下檢測,記錄色譜圖,色譜數據導入《中藥色譜指紋圖譜相似度評價系統2.0 版》軟件,采用中位數法,以S1 批樣品作為參照譜進行指紋匹配,生成共有模式指紋圖譜(圖1),可見共有10 個共有峰。相似度結果見表2,13 批多花黃精的指紋圖譜相似度在0.781~0.945 之間,其中S1(安徽省金寨縣)、S3(湖南省祁陽縣)、S8(貴州省赤水市)、S11(福建省建寧縣)和S12(江西省安福縣)5 批樣本相似度大于0.90,其它樣品低于0.90,占61.5%,這一結果與黃精P.sibiricumRed.和滇黃精P.kingianumColl.et Hemsl.的HPLC 指紋圖譜研究結果不一致[22?23],其原因是否是產地或生長年限等影響了其多糖組成,還有待進一步深入研究。本研究中采集自湖南省慈利縣的S13 號相似度最低為0.781,觀察發現與其他樣品相比,S13 號樣品根莖較幼嫩,已有研究表明黃精幼嫩部位的多糖含量較成熟部位低[29],這可能是其相似度較低的原因。
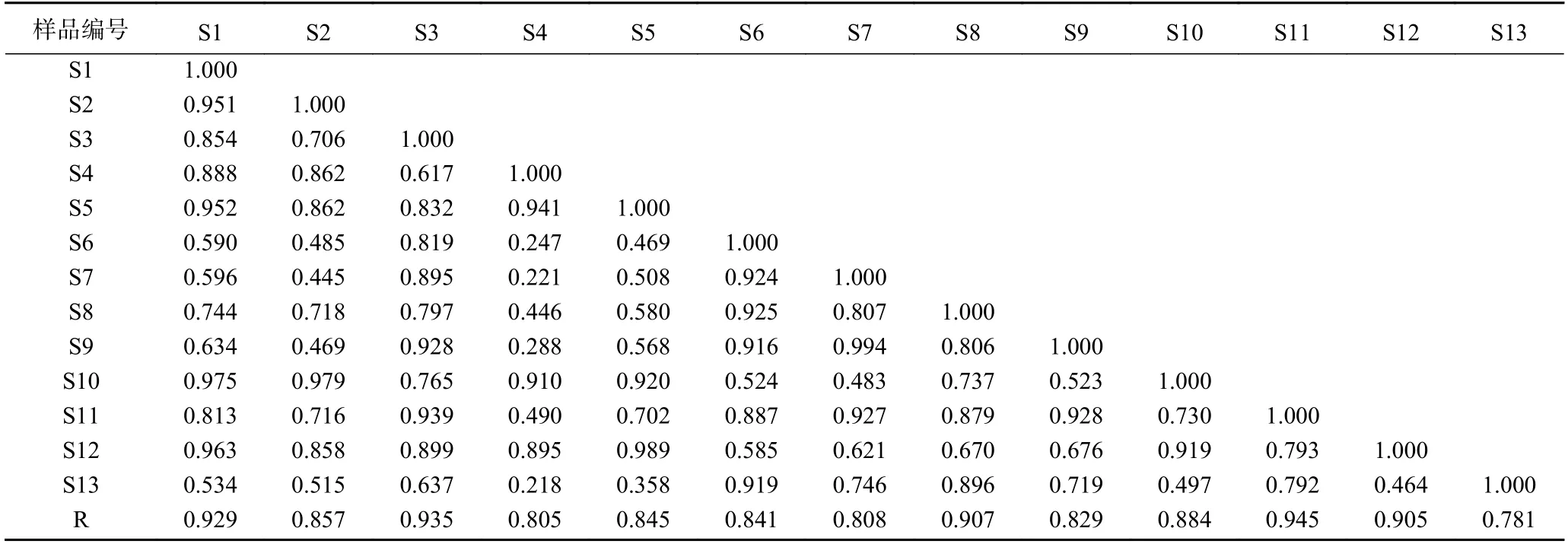
表2 13 批不同產地多花黃精相似度Table 2 Similarity evaluation of 13 batches of P.cyrtonema from different localities
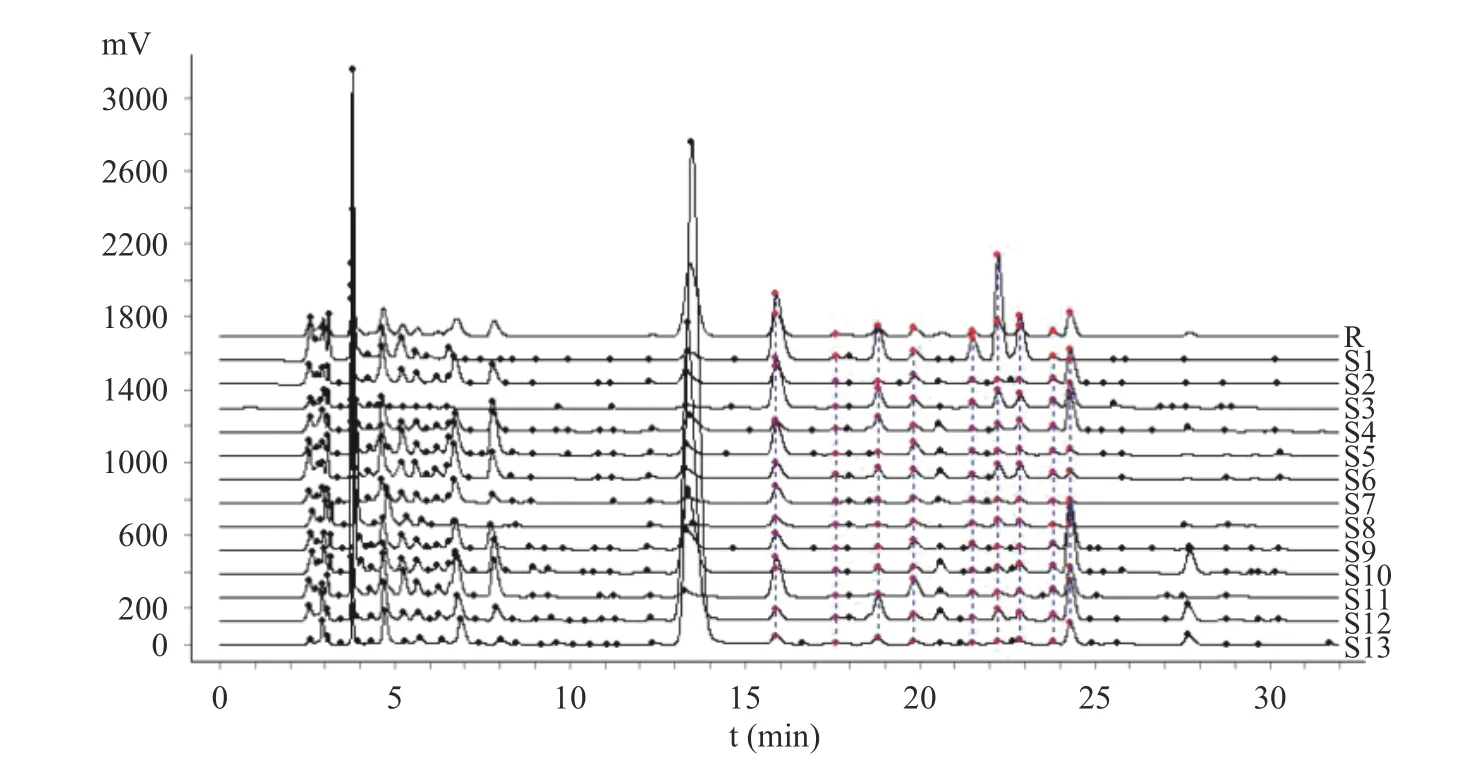
圖1 13 批多花黃精多糖柱前衍生化HPLC 指紋譜Fig.1 Pre-column derivation HPLC characteristic fingerprint of polysaccharide hydrolysate from 13 batches of P.cyrtonema
2.4 單糖定性鑒定
樣品及單糖對照品的PMP-HPLC 色譜圖見圖2。通過與對照品的保留時間比對,發現10 個共有峰中,5 個與單糖對照品保留時間一致,即4 號峰(18.80 min)為半乳糖醛酸,6 號峰(20.13 min)為葡萄糖醛酸,7 號峰(21.50 min)為半乳糖,8 號峰(22.22 min)為葡萄糖,9 號峰(22.84 min)為木糖,單糖研究結果與王坤等[30]對多花黃精單糖組成研究結果基本一致。在前期研究中,何連軍等[14]采用高效陰離子交換色譜-脈沖安培檢測法,檢測到多花黃精含有果糖,而本研究未檢測到果糖,主要原因是由于PMP 僅能與醛糖發生衍生化反應,果糖屬于酮糖,不發生衍生反應[31]。
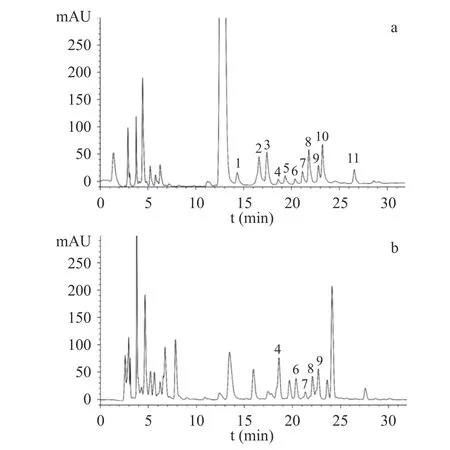
圖2 多花黃精藥材共有模式圖譜及對照品HPLC 圖Fig.2 HPLC fingerprints of P.cyrtonema and chromatograms of reference substances
2.5 總多糖含量
多糖是黃精藥材的重要質量標志物[32],按2020年版《中華人民共和國藥典》含量測定項規定,其干燥品含多糖以無水葡萄糖(C6H12O6)計,不得少于7.0%。本研究顯示,13 批多花黃精藥材的多糖含量符合要求,在7.18%~16.27%范圍內,結果見表3。可見總多糖含量最高的S3 號(湖南省祁陽縣),最低是S9 號(湖南省懷化市),約相差1 倍,表明不同產地多花黃精多糖含量有明顯差異,與相似度分析結果一致。已有研究表明,產地和生長年限是影響黃精多糖含量的重要影響因素[29,33],本研究中,樣品為隨機采集,產地和生長年限可能是形成其多糖差異的原因。
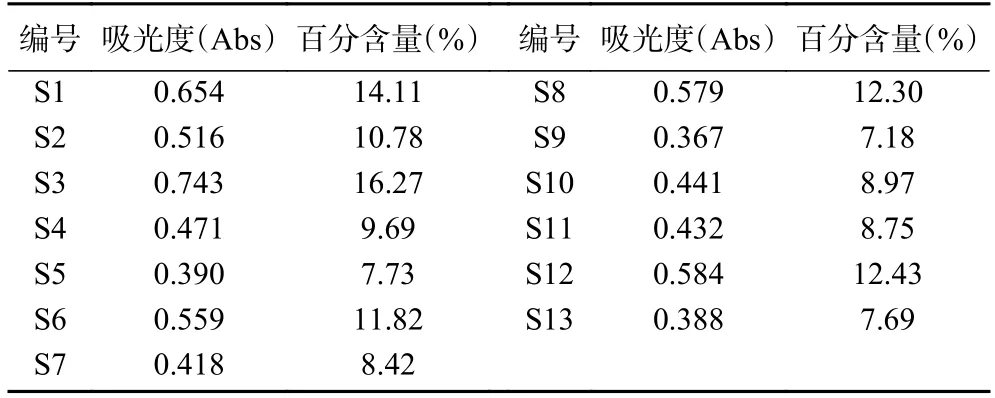
表3 13 批多花黃精的總多糖含量測定結果Table 3 Determination results of total polysaccharide content of 13 batches of P.cyrtonema
2.6 聚類分析
聚類分析(HCA)是按照個體數據特征對樣本進行分類的一種方法,其同一類別個體間具有較高的相似度[11]。將13 批樣品單位質量藥材峰面積進行量化,得到13×10 階的數據矩陣,采用SPSS 20.0 版軟件,瓦爾德法(Ward)進行分析,結果見圖3。可見當判別條件距離為10 時,13 批多花黃精藥材被分成兩類,I 類包括12 個樣品,為S1~S12 號,II 類僅有S13 號(湖南省慈利縣),若以相似度0.80 為界限,聚類結果與相似度和多糖含量結果一致。不同產地的樣品在聚類圖上無明顯區分,表明栽培多花黃精藥材的多糖成分類別差異不大,S13 號樣本偏離的原因可能是采收年限、種質資源或栽培技術差異造成的[34]。

圖3 不同產地多花黃精系統聚類分析Fig.3 The cluster analysis of P.cyrtonema from different localities
2.7 主成分分析
主成分分析(PCA)是將多維具有相關性的數據壓縮為少數幾個相互獨立數據的統計方法,其在不損失主要信息的前提下實現降維,擴大樣本之間的差異,可解決由于中成藥成分復雜所致的普帶重疊分析困難[35]。為了更加全面、系統地分析13 批多花黃精之間的差異,揭示其內在關系,將上述13×10 數據矩陣采用SPSS 20.0 軟件計算特征值和方差貢獻率,見表4,以主成分的特征值大于1 和累計方差貢獻率大于85%,作為選擇主成分因子依據[36],結果顯示前三個主成分可代表89.364%的信息量。采用SIMCA-P13.0 計算PCA 得分圖,結果見圖4,13 個樣品被分成兩類,其中采集于湖南省慈利縣的S13 號樣品單獨聚為一類,與HCA 結果一致,觀察發現與其他樣品相比,S13 號樣本較為幼嫩,可能是其單獨聚為一支的原因。
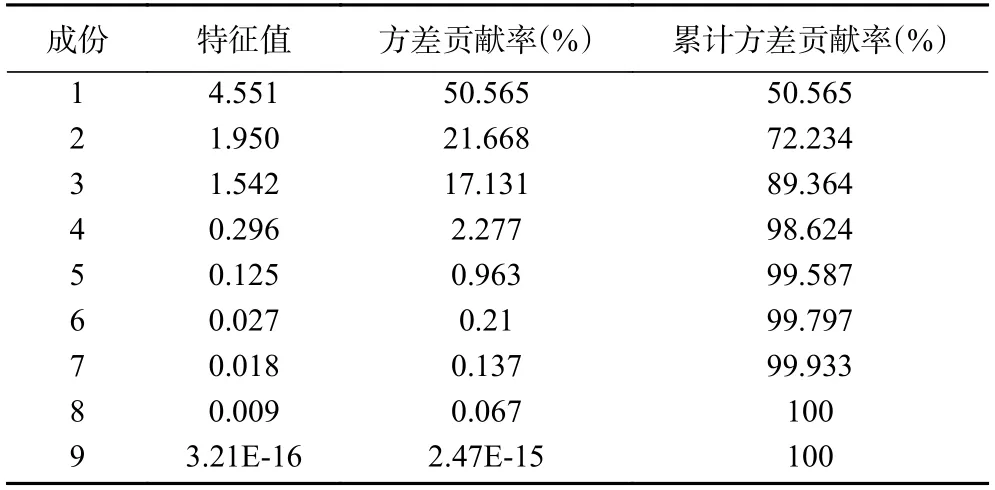
表4 特征值和方差貢獻率Table 4 Characteristic value and variance contribution rate
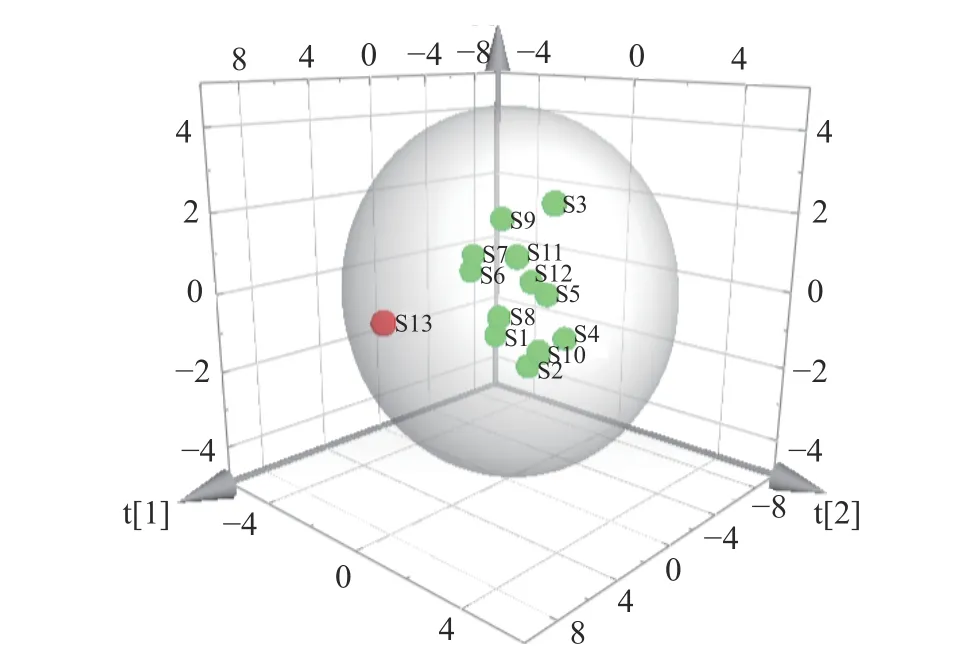
圖4 多花黃精PMP-HPLC 圖譜PCA 結果Fig.4 PCA results of PMP-HPLC fingerprints of P.cyrtonema
3 結論
本實驗首次建立了不同產地多花黃精中多糖成分的PMP-HPLC 指紋圖譜,并對其單糖組成進行了研究,在此基礎上,結合指紋圖譜技術,采用相似度分析,聚類分析和主成分分析對其指紋圖譜進行了研究,并對其總多糖含量進行了測定。實驗結果表明,13 批多花黃精單糖組成均含半乳糖醛酸、葡萄糖醛酸、半乳糖、葡萄糖、木糖,其總多糖含量范圍在7.18%~16.27%之間,HCA 和PCA 分析結果一致,樣本被分為2 類,其次,13 批多花黃精的指紋圖譜相似度較低,在0.781~0.945 之間,其中相似度低于0.90 的樣品占所研究樣品的61.5%,從一定程度反映了多花黃精藥材質量的不均一,這一結果與黃精其他基原物種研究結果不一致[22?23],其原因可能是種質資源、產地或生長年限等影響了其多糖生成,還有待進一步深入研究。綜合產地與指紋圖譜相似度、總多糖含量、單糖組成等分析,未發現明顯的規律,已有研究表明,多花黃精不同齡節間的有效成分含量差異較大[37],不同生長年限的黃精藥材其多糖成分亦存在一定差異[29],提示采收年限和栽培技術可能是造成多花黃精品質差異的原因。因此,為確保黃精藥材臨床用藥的有效和安全,有必要建立多花黃精的規范化栽培技術體系,以確保多花黃精藥材品質的一致性。
