熒光納米材料YPO4∶Sm3+@YPO4@聚乙二醇的構筑及其熒光性能
2022-07-12 07:39:56劉叢林吳錦繡賈慧靈柳召剛胡艷宏齊源昊王忠志內蒙古科技大學材料與冶金學院包頭0400
無機化學學報 2022年7期
劉叢林 吳錦繡*, 賈慧靈 柳召剛胡艷宏 王 昕 齊源昊 王忠志(內蒙古科技大學材料與冶金學院,包頭 0400)
(2輕稀土資源綠色提取與高效利用教育部重點實驗室,包頭 014010)(3內蒙古自治區稀土濕法冶金與輕稀土應用重點實驗室,包頭 014010)(4內蒙古科技大學機械工程學院,包頭 014010)(5包頭稀土研究院,包頭 014010)
0 引 言
稀土摻雜的磷酸鹽作為發光材料的優點在于吸收能力強、轉換率高,特別是在可見光區域具有很強的發射能力,且物理化學性質穩定[1-2]。稀土Sm3+離子在近紫外和可見光區存在豐富的能級,對近紫外和藍光具有強烈的光吸收性能,并可發射橙紅色熒光。Sm3+離子的光吸收范圍與InGaN芯片發射的近紫外光相匹配[3-4],其紅色發光譜線比Eu3+更豐富[5]。Sm3+離子摻雜磷酸釔(YPO4∶Sm3+)由于其優良的發光性能,作為紅色熒光粉可廣泛應用于高壓汞燈和彩色電視機等[6]。近年來,YPO4∶Sm3+納米粒子因其在高分辨率顯示器、藥物傳遞系統和生物熒光標記等方面的潛在應用而受到了極大的關注[7-8]。納米材料與微米材料相比,具有更高的比表面缺陷,導致其熒光性能下降[9]。因此提高納米材料的發光性能是當今研究領域的主要問題。
Zhu等[10]利用水熱法合成了具有增強光致發光作用的 YVO4∶Eu3+@YPO4核殼結構,通過調配 YVO4∶Eu3+和YPO4的最佳物質的量之比,使其發光強度提高了44%。吳錦繡[11]利用微波法合成了YPO4∶Sm3+@CS球形熒光粉,表面修飾后的產物具有較好的水溶性和生物相容性。Lim等[12]利用水熱法制得LaPO4∶Er3+@LaPO4核殼納米顆粒,其納米雜化膜的發光強度和壽命得到顯著提高。Hu[13]提出了在YPO4∶Sm3+晶體中引入Tb3+離子以改善其發光性能,使Sm3+在室溫下的發光強度和持續時間提高了約14倍。Ansari[14]在低溫條件下制備了單分散、球形、高水溶性的熒光納米材料GdPO4∶Eu3+@LaPO4@SiO2,并且包覆后的產物發光性能顯著提高,表明LaPO4層包覆成功。但是關于構筑雙層核殼結構的納米熒光粉 YPO4∶Sm3+@YPO4@PEG(聚乙二醇)還未見相關文獻報道。
綜上可知,選用合適的材料對納米發光材料進行表面包覆和修飾,不僅可以改善納米材料的疏水性能,還可以提高其熒光性能。主要是由于表面包覆層可以通過抑制能量轉移過程的能量損失來提高納米熒光粉的發光性能[15-18]。本課題組已經研究了球形納米熒光粉YPO4∶Sm3+的形貌形成機理,探究了該類熒光粉的結構和形貌對發光性能的影響[19],同時也研究了不同酸度對球形納米熒光粉YPO4∶Sm3+的結構和形貌及其發光性能的影響[20]。在此基礎上,先用同質晶體YPO4包覆球形納米熒光粉YPO4∶Sm3+,得到核殼結構的熒光納米材料YPO4∶Sm3+@YPO4。然后用PEG進行外層包覆,得到雙層核殼結構的熒光納米材料YPO4∶Sm3+@YPO4@PEG。該雙層熒光納米材料不僅具有親水性和生物相容性,同時也增強了納米熒光粉YPO4∶Sm3+的熒光性能。
1 實驗部分
1.1 試劑
Y2O3、濃磷酸、濃硝酸和NaOH(AR)購自國藥集團化學試劑有限公司,Sm2O3(99.999%)購自包頭稀土研究院,PEG(Mw=1 000)購自國藥集團化學試劑有限公司。試驗用水全部為去離子水。
1.2 YPO4∶Sm3+的制備
參照文獻[15-16]提供的方法制備YPO4∶Sm3+熒光粉,具有步驟如下:分別用移液管量取2 mL Sm(NO3)3(0.05 mol·L-1)溶液和 9.8 mL Y(NO3)3(0.5 mol·L-1)溶液置于50 mL燒杯中,將燒杯放置在磁力攪拌器上,在持續攪拌的情況下逐滴加入10 mL濃度為1.5 mol·L-1的磷酸溶液。滴加完畢后繼續攪拌10 min。攪拌結束后使用6 mol·L-1的NaOH溶液調節其pH約為3。繼續攪拌10 min后超聲分散20 min,之后將反應體系移入高壓反應釜的聚四氟乙烯容器中,填充度為80%,隨后將高壓反應釜置于鼓風干燥箱中,200℃反應12 h。反應結束后,反應釜隨爐冷卻到室溫。最后通過離心、洗滌、干燥和研磨等步驟得到產物YPO4∶Sm3+,并稱重后封裝。
1.3 YPO4∶Sm3+@YPO4的制備
分別制備 YPO4∶Sm3+(核)、YPO4(包覆劑)物質的量之比為 5∶1、6∶1、8∶1 和 9∶1 的熒光材料 YPO4∶Sm3+@YPO4。以6∶1的材料制備過程為例:首先稱取0.3 g YPO4∶Sm3+和15 mL無水乙醇置于瑪瑙研缽中研磨,使其充分分散,然后將反應物移入200 mL燒杯,并加入15 mL去離子水,在超聲振動器中振動分散 30 min。量取 1.8 mL Y(NO3)3(0.1 mol·L-1)溶液和8.2 mL水于該燒杯中,充分攪拌混合,然后量取5.4 mL磷酸(1.5 mol·L-1)溶液逐滴加入該燒杯中。最后用濃度為3 mol·L-1的NaOH調節pH約為3。攪拌30 min后將反應體系移入高壓反應釜的聚四氟乙烯容器中,隨后將高壓反應釜置于鼓風干燥箱中,200℃反應12 h。反應結束后,隨爐冷卻到室溫。最后通過離心、洗滌、干燥和研磨等步驟得到產物YPO4∶Sm3+@YPO4并稱重后封裝。后文如無特別指出YPO4∶Sm3+@YPO4均指物質的量之比為6:1時制備的產物。
1.4 YPO4∶Sm3+@YPO4@PEG的制備
稱取0.2 g上述制備的YPO4∶Sm3+@YPO4置于瑪瑙研缽中,加入少量無水乙醇后研磨,將研磨好的產物倒入250 mL燒杯中,加入100 mL去離子水超聲分散30 min,得到2 g·L-1懸浮液A。將0.2 g PEG溶于100 mL去離子水中,得到2 g·L-1PEG溶液B。將懸浮液A移入500 mL的四口圓底燒瓶中,在微波反應器中60℃加熱。將溶液B以每滴9~10 s的速度滴入不斷攪拌的懸浮液A中得到反應體系C。C在60℃下微波反應7.5 h。待反應結束,C冷卻到室溫后,將其移入500 mL燒杯中并加入50 mL正己烷常溫陳化12 h。最后,將C進行離心、洗滌和干燥,獲得最終產物YPO4∶Sm3+@YPO4@PEG,并研磨和封裝。
1.5 樣品表征
用D8 ADVANCE X射線粉末衍射儀(XRD,工作電壓40 kV,工作電流30 mA,德國Bruker)對產物進行物相分析,采用CuKα(λ=0.154 05 nm)輻射,掃描范圍 10°~80°,掃描速度 4(°)·min-1。用 Sigma500 AMCS場發射掃描電子顯微鏡(SEM,工作電壓3 kV,德國卡爾蔡司)和JEOL 2100F透射電子顯微鏡(TEM,工作電壓200 kV,德國Breker)測定產物的元素組成、形貌、尺寸等。用Alpha傅里葉變換紅外光譜儀(FT-IR,德國BROKER)測試樣品的紅外光譜,采用KBr壓片。用F-4600熒光分光光度計(FL,日本日立高新技術集團)測定樣品的激發和發射光譜。用FL920系列穩態/瞬態熒光光譜儀(FL,英國愛丁堡公司)測量樣品熒光壽命。以上測試均在室溫下進行。采用美國TA Instruments儀器公司生產的熱重(TG)分析儀(型號為SDTQ600)測試產物的熱穩定性和熱分解過程。
2 結果與討論
2.1 結構和形貌分析
利用Jade 6.0軟件分析了包覆前后產物的XRD圖(圖1)可知,圖中所有衍射峰都與YPO4標準卡片(PDF No.83-0658)的衍射峰相對應,沒有出現其它雜峰,表明所制備的產物為純相,具有四方晶系結構,其空間點陣為I41/amd,晶胞參數:a=0.688 2 nm,c=0.601 8 nm,Z=4,最強衍射峰為(200)晶面。通過相關軟件分析計算了(200)晶面的結晶度,YPO4∶Sm3+@YPO4@PEG的結晶度為72.47%,YPO4∶Sm3+@YPO4的結晶度為78.85%,YPO4∶Sm3+的結晶度為78.65%,說明PEG包覆后的產物在(200)晶面的結晶度降低,這主要是由于PEG為非晶態。由此可以推斷PEG和YPO4包覆不會引起產物晶體結構的變化[21-22]。
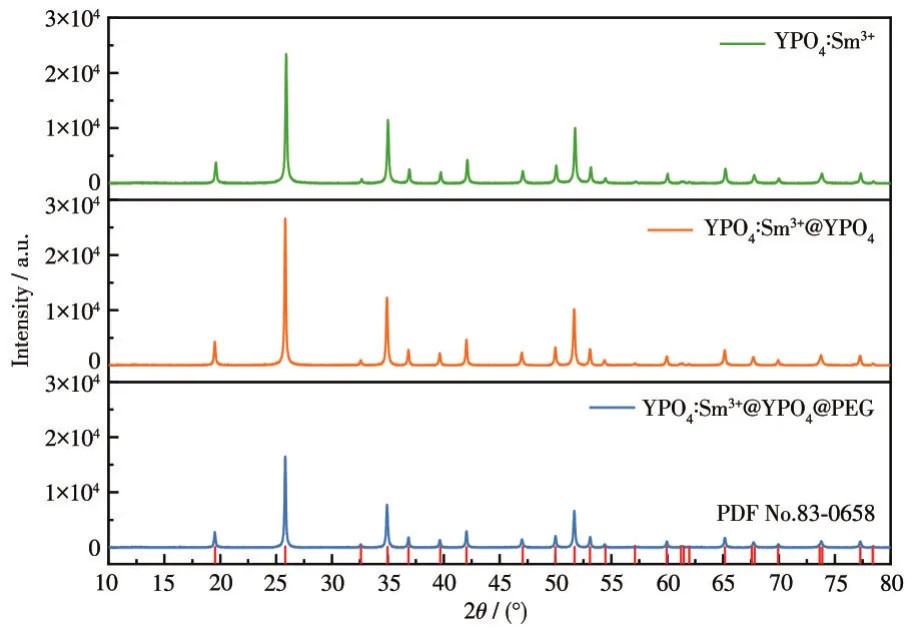
圖1 YPO4∶Sm3+包覆前后的XRD圖Fig.1 XRD patterns of YPO4∶Sm3+before and after coating
由圖2的SEM圖可知,包覆前后的產物形貌沒有太大的變化,都呈球形。用粒度分析軟件Nano measurer 1.2算出平均粒徑。分析可知包覆前產物的平均粒徑為60~80 nm;無機物YPO4包覆的產物平均粒徑為65~90 nm。納米粒子是由晶粒部分團聚形成,由于晶粒受表面效應影響,粒子間有相互聚集的趨勢[23]。有機PEG包覆產物YPO4∶Sm3+@YPO4@PEG的平均粒徑為70~100 nm。可知包覆層的厚度為10~20 nm。由圖可知,PEG包覆后的產物顆粒表面光滑、圓潤、分散性較好[24-25]。由此可知,選擇PEG作為包覆劑可以補充納米核的表面缺陷。

圖2 YPO4∶Sm3+包覆前后的SEM圖Fig.2 SEM images of YPO4∶Sm3+before and after coating
圖3為YPO4包覆前后的能譜(EDS)圖。從圖中可知,納米發光材料 YPO4∶Sm3+和 YPO4∶Sm3+@YPO4的組成元素都僅為O、Y、Sm和P,這說明YPO4包覆前后的產物元素組成相同,并沒有出現其它雜質。
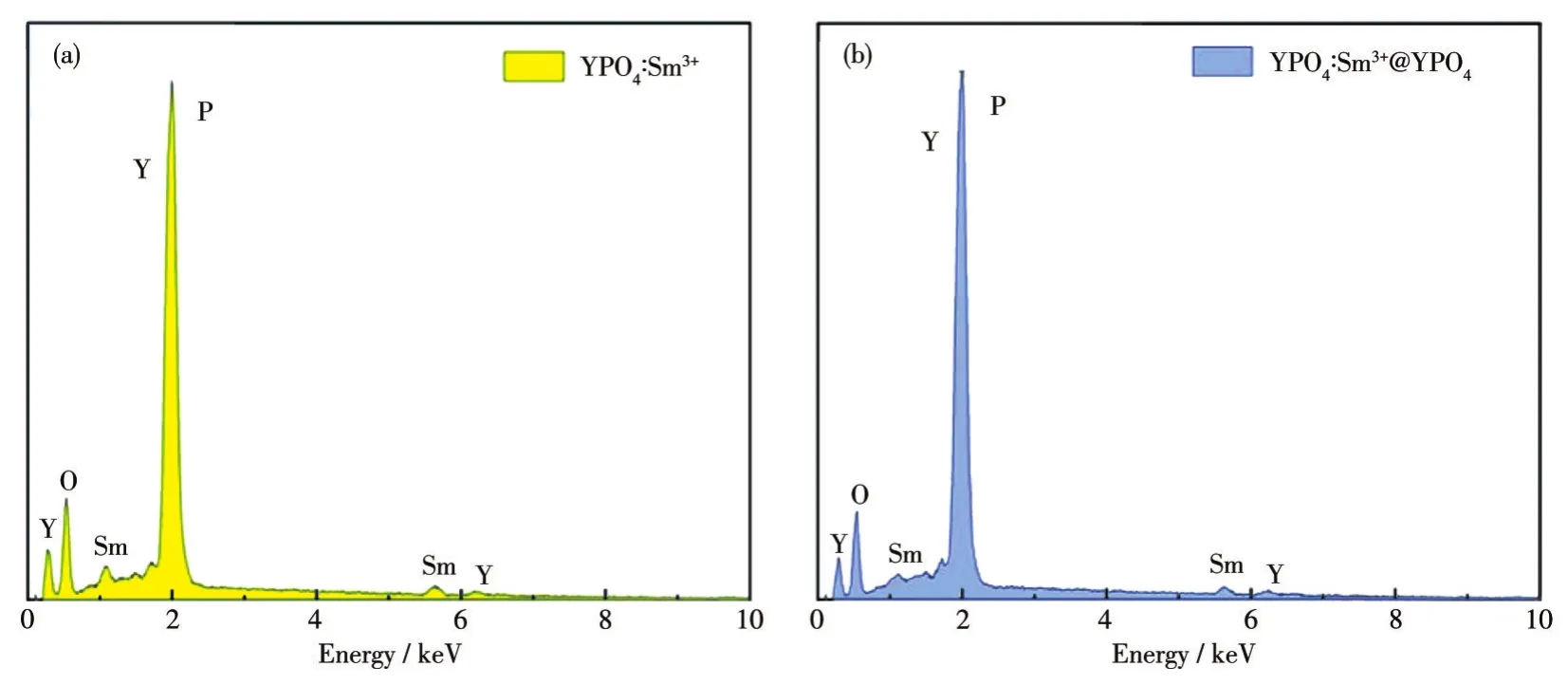
圖3 YPO4∶Sm3+包覆前 (a)后 (b)的EDS譜圖Fig.3 EDS spectra of YPO4∶Sm3+before(a)and after(b)coating
為了進一步研究包覆層的厚度,對包覆后的產物進行了TEM測試。YPO4∶Sm3+@YPO4顆粒平均粒徑為65~90 nm(圖4),YPO4∶Sm3+@YPO4@PEG的平均粒徑為 70~100 nm(圖5),YPO4∶Sm3+的平均粒徑為60~80 nm[20],可知內層包覆厚度為5~10 nm,外層包覆厚度為5~10 nm,這與SEM分析結果基本吻合。以上結果進一步證明YPO4和PEG都成功包覆在納米熒光粉YPO4∶Sm3+的表面[26]。從高分辨透射電鏡(HRTEM)圖中可知 YPO4∶Sm3+@YPO4的(200)晶面間距為 0.244 2 nm,YPO4∶Sm3+@YPO4@PEG的(312)晶面間距為0.190 7 nm,這與標準卡片(PDF No.83-0658)對應的晶面間距相吻合。選區電子衍射(SAED)圖中由內向外所表示的晶面指數符合XRD分析結果。YPO4∶Sm3+@YPO4的電子衍射花樣呈現明亮、清晰的圓環,說明產物是晶體。而PEG包覆后YPO4∶Sm3+@YPO4@PEG的電子衍射光環較為彌散,這與XRD分析的結果相吻合,這主要是由于非晶態PEG表面包覆層降低了圖像的清晰度[27]。

圖4 YPO4∶Sm3+@YPO4的(a)TEM圖、(b)HRTEM圖、(c)SEAD圖Fig.4 (a)TEM image,(b)HRTEM image,and(c)SEAD image of YPO4∶Sm3+@YPO4

圖5 YPO4∶Sm3+@YPO4@PEG的(a)TEM圖、(b)HRTEM圖、(c)SEAD圖Fig.5 (a)TEM image,(b)HRTEM image,and(c)SEAD image of YPO4∶Sm3+@YPO4@PEG
2.2 紅外光譜分析
從圖6a中可以看出位于1 200~985 cm-1處的寬吸收帶歸屬于P—O—P鍵反對稱伸縮振動,在660 cm-1處的吸收帶為P—O鍵的反對稱彎曲振動,并且在520 cm-1處的吸收帶歸屬于Y—O和Sm—O的重合振動。在3 486和1 655 cm-1處有2個吸收帶,分別歸屬于水的H—O鍵彎曲和伸縮振動吸收峰,這可能是在壓片過程中溴化鉀中的水或產物表面吸附的水[28-29]所致。在2 923、2 852和2 026 cm-1處存在弱的吸收帶,可能是在測試的過程中產物吸收了空氣中的二氧化碳導致的。從圖6a中可以看出,包覆前后產物的紅外光譜較為相近,但YPO4∶Sm3+@YPO4@PEG 相比于 YPO4∶Sm3+在 1 655 cm-1處的H—O藍移了10 cm-1,說明該H—O歸屬于PEG分子中的羥基,也有可能是稀土離子Y與羥基中的O形成Y—O鍵。由圖6b可知,在3 389和1 630 cm-1處的吸收帶歸屬于吸附水分子和PEG的羥基振動吸收;位于1 470cm-1處的吸收帶屬于PEG的彎曲振動吸收;在2 879、964和842 cm-1處的吸收帶歸屬于PEG分子的CH2振動吸收[30-32]。位于1 118 cm-1處的吸收帶是PEG的C—O—C鍵的伸縮振動峰。YPO4∶Sm3+@YPO4@PEG的紅外光譜圖與PEG相差較大。但是在2 879、964和842 cm-1處有微弱的峰,歸屬于PEG分子中CH2振動吸收,說明PEG包覆在YPO4∶Sm3+@YPO4的表面。PEG中的羥基與YPO4∶Sm3+@YPO4中的稀土離子(R3+)配位形成O—R3+鍵[33-34],該鍵與YPO4∶Sm3+在530~630 cm-1處的吸收帶與 Y—O鍵和Sm—O鍵的振動相重合。通過以上分析可知,PEG成功包覆在YPO4∶Sm3+@YPO4的表面。
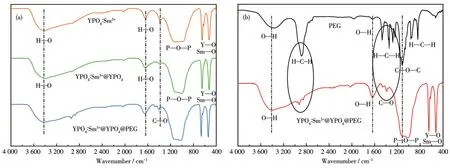
圖6 YPO4∶Sm3+包覆前后及PEG的紅外光譜圖Fig.6 Infrared spectra of YPO4∶Sm3+before and after coating and PEG
2.3 TG分析
圖7為YPO4∶Sm3+包覆前后的TG圖。從圖中可知,包覆前后樣品的TG曲線在700~1 000℃范圍內趨于平緩,說明該類熒光粉在700℃比較穩定。其中 YPO4∶Sm3+失重率為 4.2%,YPO4∶Sm3+@YPO4失重率為6.0%,相較于YPO4∶Sm3+失重率增加了1.8%,這可能是因為在YPO4∶Sm3+表面進行同質晶體YPO4的外延包覆后表面吸附的結晶水增加,導致失重率略有增加。YPO4∶Sm3+@YPO4@PEG在0~340℃范圍內由于結晶水分解導致的失重率為2%。在340~420℃范圍內有明顯的失重現象,此時是包覆層PEG的分解過程,在420℃時PEG完全分解[35],失重率為4.5%。PEG包覆后樣品總失重率為6.5%,相對于包覆前明顯增加。這進一步證明了YPO4和PEG成功包覆在YPO4∶Sm3+表面。
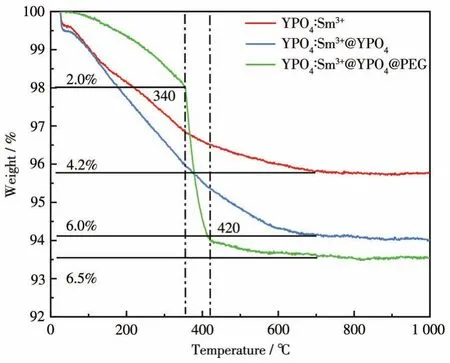
圖7 YPO4∶Sm3+包覆前后的TG曲線對比Fig.7 Comparison of TG curves for YPO4∶Sm3before and after coating
2.4 包覆機理
通過Materials Studio軟件繪制了YPO4∶Sm3+@YPO4@PEG包覆機理示意圖(圖8)。圖8a為YPO4∶Sm3+的晶體結構圖,圖8b表示YPO4包覆后的三維立體圖。圖8c為PEG單體分子結構示意圖。由紅外光譜圖可知,PEG中的羥基與稀土離子配位形成O—R3+鍵(圖8d)。PEG單體分子中有2個羥基,其中一個羥基與YPO4∶Sm3+@YPO4中的稀土離子成鍵,另一個羥基在產物外層,因此YPO4∶Sm3+@YPO4@PEG具有親水性和生物相容性。圖9為YPO4包覆前后的宏觀示意圖,YPO4∶Sm3+表面內層包覆無機物YPO4,外層包覆有機物PEG,得到雙層核殼結構的YPO4∶Sm3+@YPO4@PEG。
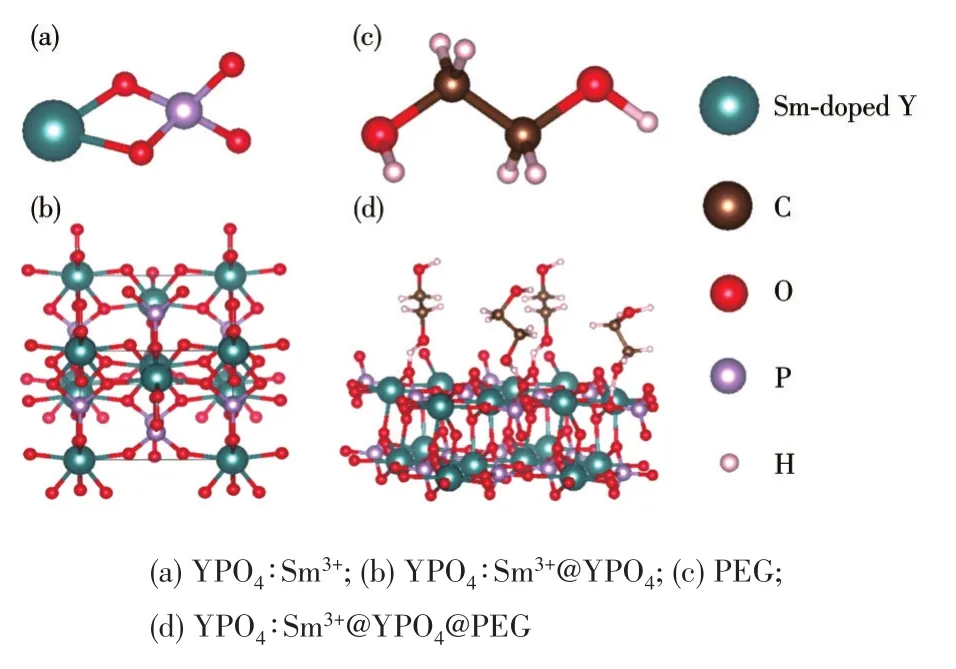
圖8 包覆機理微觀示意圖Fig.8 Microscopic schematic diagram of coating mechanism
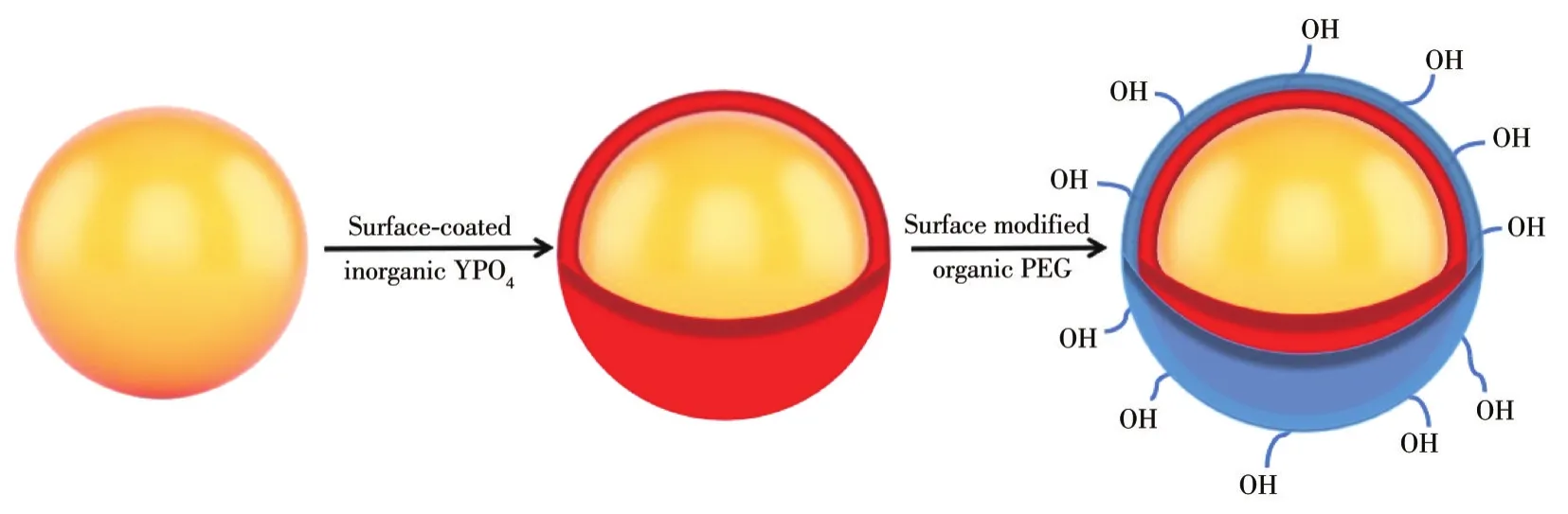
圖9 包覆機理宏觀示意圖Fig.9 Macroscopic schematic diagram of coating mechanism
2.5 熒光性能分析
圖10是包覆前后熒光納米材料的激發和發射光譜圖。由圖10可知,包覆前后激發峰和發射峰的位置基本沒有改變,但是熒光強度明顯增強。由圖10a可知,在404 nm可見光的激發下,Sm3+的4個主要特征發射峰分別位于564、603、648和 705 nm[36],并且在603 nm處的熒光強度最強。從圖10b中可以看出,在603 nm波長檢測下,產物在300~500 nm范圍內有一系列的激發峰,均是Sm3+的4f組態內的f-f躍遷,其中在404 nm處出現最強激發峰。這些激發峰歸屬于 Sm3+的6H5/2→4K17/2(346 nm)、6H5/2→4H7/2(363 nm)、6H5/2→4P7/2(376 nm)、6H5/2→4P3/2(404 nm)、6H5/2→4P5/2(418 nm)、6H5/2→4G9/2(441 nm)、6H5/2→4I13/2(466 nm)和6H5/2→4I11/2(478 nm)能級躍遷[37],可見納米熒光粉YPO4∶Sm3+可被紫外和可見光有效激發。
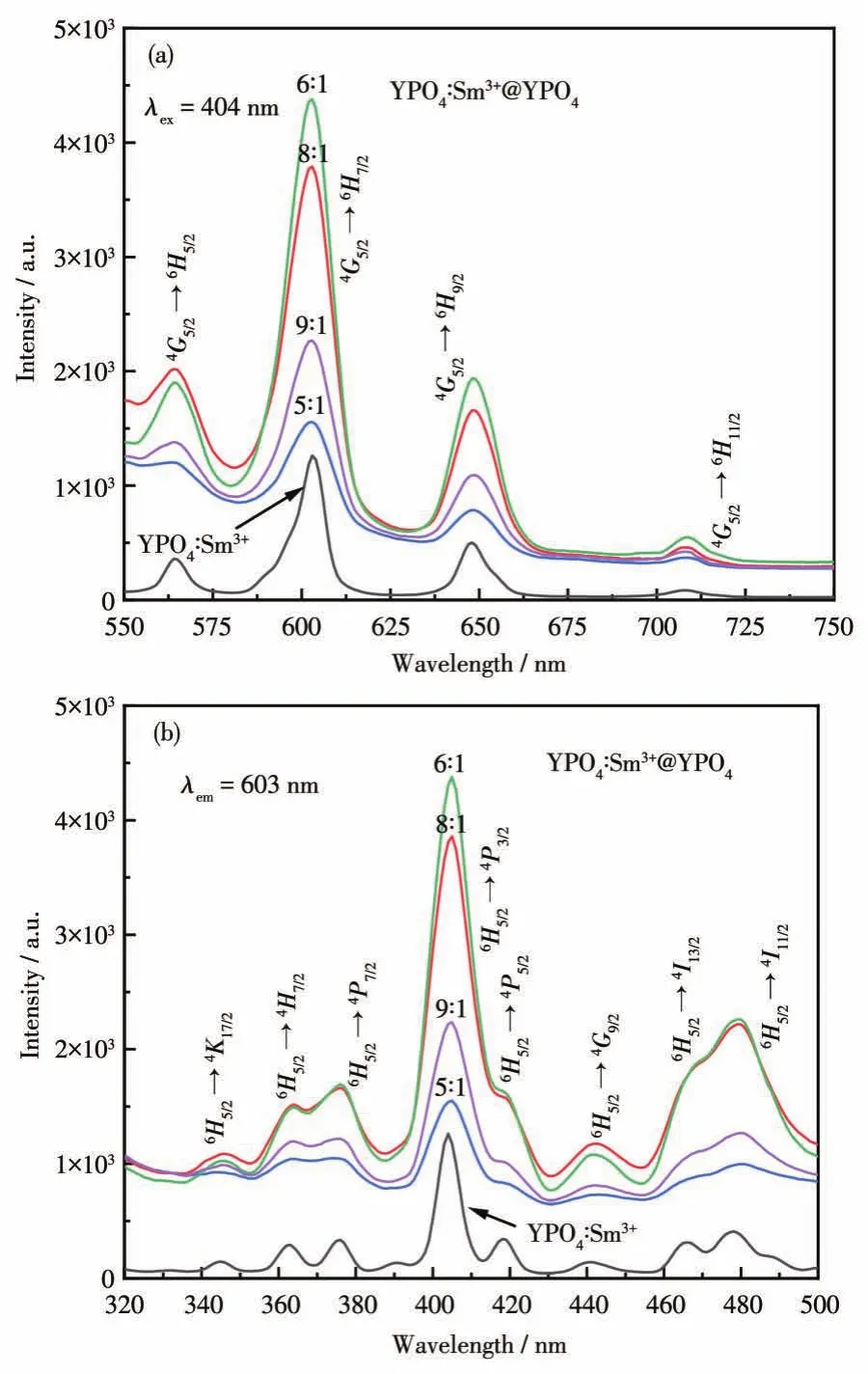
圖10 YPO4∶Sm3+和 YPO4∶Sm3+@YPO4發射 (a)和激發(b)光譜圖Fig.10 Emission(a)and excitation(b)spectra of YPO4∶Sm3+and YPO4∶Sm3+@YPO4
與其它的異質結構相比,YPO4∶Sm3+與包覆劑YPO4有相同的晶體結構和單胞參數,因而更容易外延生長到YPO4∶Sm3+納米晶體上[10,15]。當二者物質的量之比為5∶1和9∶1時,熒光強度增強了約0.5和1倍;當二者物質的量之比為8∶1時,熒光強度增強了2倍多。當二者物質的量之比為6∶1時,熒光強度增強了3倍多。說明核YPO4∶Sm3+與包覆劑YPO4的物質的量之比為6∶1時包覆層厚度最佳,表面復合率、表面缺陷密度和表面態密度都大大降低,有效地減少了非輻射衰變通道,有利于能量的轉換,從而使熒光強度達到最高[38]。并且O—R3+鍵的數量增多,O2-離子的2p軌道吸收的能量達到最大,傳遞給Sm3+離子的能量也最多,所以熒光強度最高[39-40]。可見表面包覆劑YPO4的包覆量及厚度會影響產物的發光強度。綜上可知,同核無機鹽YPO4表面包覆層對發光納米核起到了保護作用,這可能是通過抑制能量轉移過程的能量損失來使核的熒光性能增強[20]。
從圖11可知,包覆PEG前后產物的激發峰和發射峰的位置沒有改變,但是熒光強度明顯增強,位于603 nm處的特征峰熒光強度最高。YPO4∶Sm3+@YPO4@PEG的熒光強度比YPO4∶Sm3+增強了5倍多,較內層包覆產物YPO4∶Sm3+@YPO4增強了0.5倍多。PEG包覆后產物熒光強度增強是因為最外層包覆起到了鈍化作用,減少了晶體的缺失,也可能因為PEG作為聚合物包覆有助于將光致發光的量子產率提高[41]。
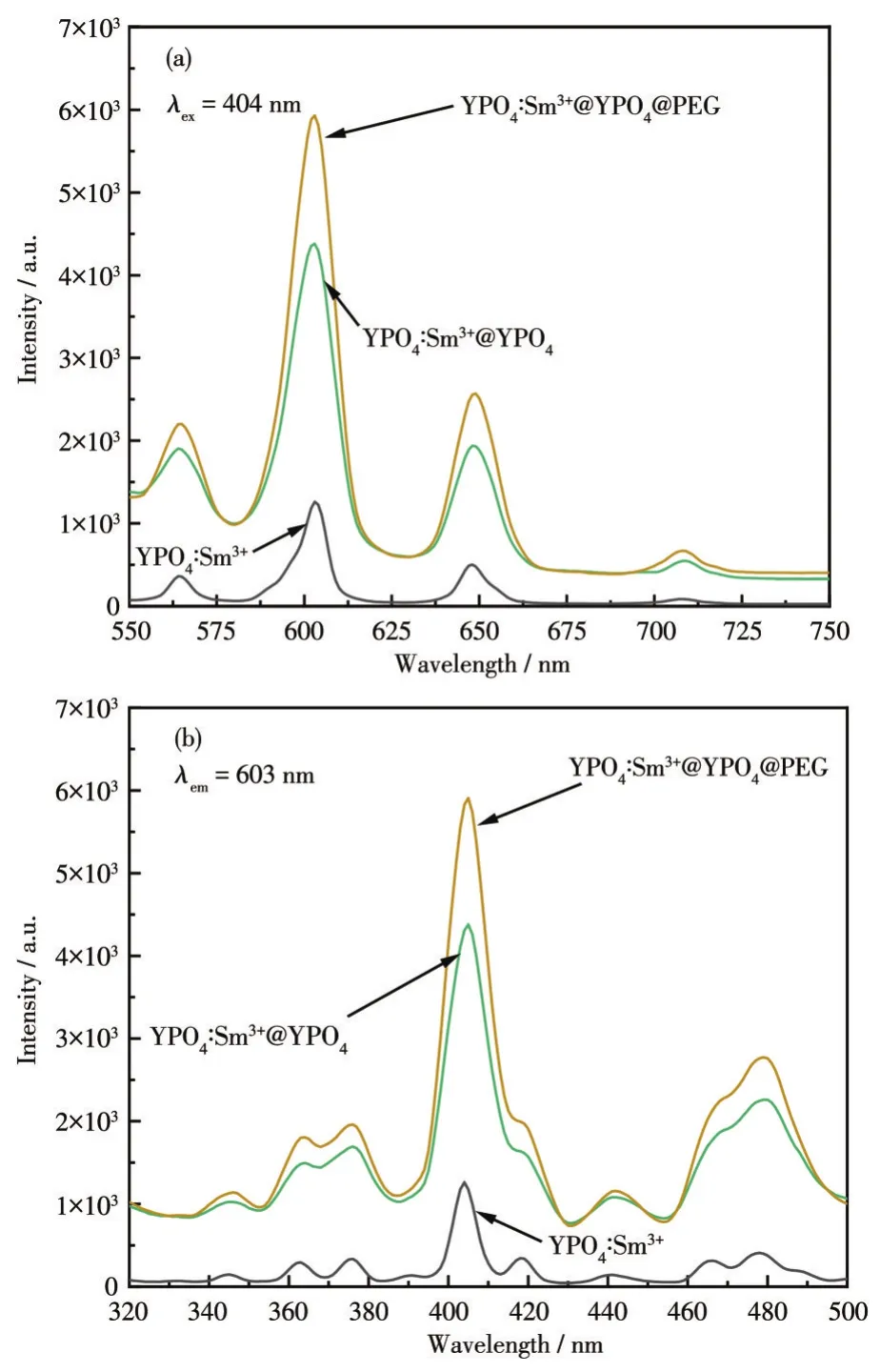
圖11 YPO4∶Sm3+@YPO4包覆前后發射 (a)和激發(b)光譜圖Fig.11 Emission(a)and excitation(b)spectra of YPO4∶Sm3+@YPO4before and after coating
發光顏色對于熒光粉的實際應用也至關重要。在本實驗中,用CIE 1931軟件計算了包覆前后的產物在404 nm近紫外光激發下發光的色坐標,并在色度圖中標注了其位置(圖12)。其中YPO4∶Sm3+的色坐標為(0.540 2,0.447 3),YPO4∶Sm3+@YPO4的色坐標為(0.566 8,0.432 3),YPO4∶Sm3+@YPO4@PEG 的色坐標為(0.580 3,0.418 9)。由此可知,包覆前后產物的色坐標略微有變化,發光顏色均為橙紅色。可見不具有發光性能的包覆層對熒光粉的發光顏色影響不大。
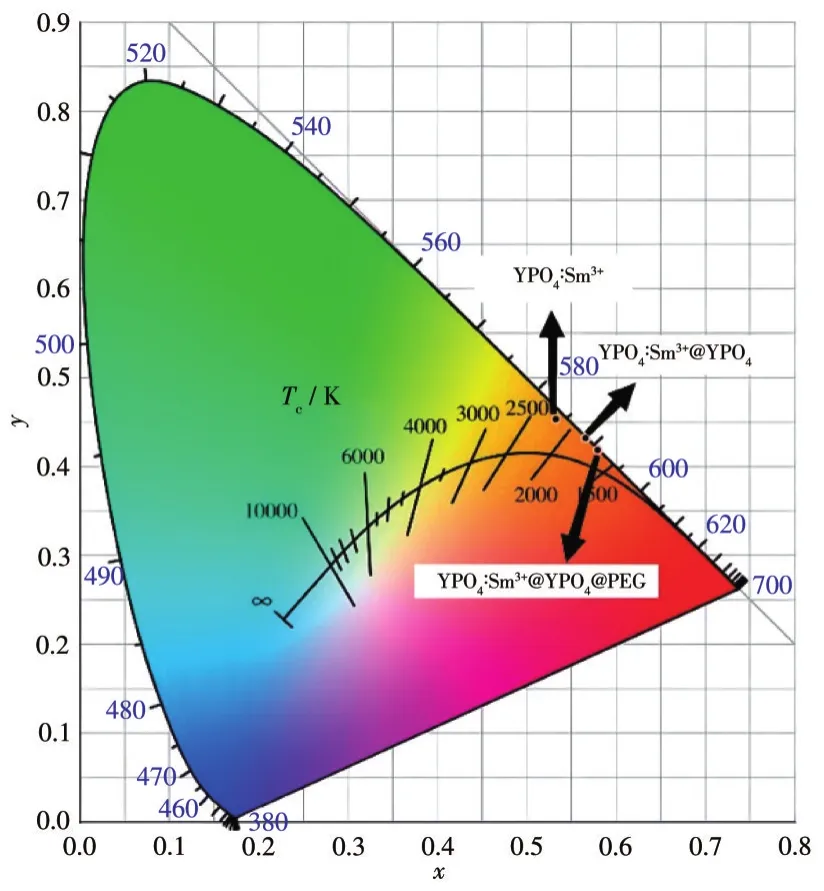
圖12 YPO4∶Sm3+包覆前后熒光發射顏色在色度圖中的位置Fig.12 Fluorescence color positions of YPO4∶Sm3+before and after coating in chromaticity diagram
2.6 熒光壽命分析
為了進一步研究包覆劑對納米熒光粉熒光性能的影響,測量了用不同產物的熒光壽命,如圖13所示。圖中為Sm3+的4G5/2→6H7/2躍遷衰減曲線,衰減曲線符合雙指數函數,使用公式1進行二次擬合[42-43]:
其中It表示在t時的熒光強度,τ1和τ2是長壽命和短壽命,A1和A2為常數。長壽命組分代表了納米粒子中大量的本征衰變時間,短壽命與表面附近的稀土離子的衰減有關,其平均壽命(τav)可以用式2[42-43]計算:

利用熒光衰減曲線計算得到 YPO4∶Sm3+、YPO4∶Sm3+@YPO4(5∶1)、YPO4∶Sm3+@YPO4(6∶1)、YPO4∶Sm3+@YPO4(8∶1)、YPO4∶Sm3+@YPO4(9∶1)的平均熒光壽命分別為2.36、2.38、2.67、2.57、2.40 ms,包覆后產物的壽命比包覆前增加了0.02~0.31 ms。可見YPO4包覆層厚度不同對熒光納米材料YPO4∶Sm3+的熒光壽命影響差別不大,這可能是測試過程中的誤差所致。
由圖13b可知,PEG包覆后的產物YPO4∶Sm3+@YPO4@PEG熒光壽命為2.37 ms,相比于YPO4∶Sm3+的熒光壽命增加了0.01ms,但是相較于YPO4∶Sm3+@YPO4的產物壽命減少了0.30ms。可能是PEG包覆后產物表面生成了保護層,減少了晶體的缺失,且產物的外層上有很多羥基裸露,增加了非輻射躍遷的速率從而縮短了輻射躍遷的壽命[41-42]。還可能是因為Sm3+之間的能量傳遞速率增大從而減少了熒光壽命,任何一種趨于和自發發射過程相競爭時都會降低激發態壽命[43]。由此可知,所有包覆前后產物的熒光壽命都在3 ms以內,與相關文獻相近[43]。
2.7 YPO4∶Sm3+@YPO4@PEG溶膠的穩定性
為了進一步驗證PEG包覆后產物的親水性,將YPO4∶Sm3+@YPO4@PEG配成濃度為 1.25 g·L-1的溶膠,在常溫下保存,每天在同一時間內測量其熒光強度,測量周期為9d。圖14為YPO4∶Sm3+@YPO4@PEG溶膠隨時間變化的熒光強度示意圖。在402 nm激發下,YPO4∶Sm3+@YPO4@PEG位于603 nm處的特征峰熒光強度達到最高。隨著測量天數增加,YPO4∶Sm3+@YPO4@PEG溶膠的熒光強度變化較小,基本趨于穩定。說明了納米熒光粉YPO4∶Sm3+@YPO4@PEG能夠在水中穩定存在9 d。通過以上分析證明了PEG包覆后的產物具有親水性和生物相容性,與理論相一致。

圖14 YPO4∶Sm3+@YPO4@PEG溶膠熒光光譜隨時間的變化Fig.14 Variations of fluorescence spectra of YPO4∶Sm3+@YPO4@PEG sol with time
3 結 論
以YPO4作為無機包覆劑,用水熱法對熒光納米材料YPO4∶Sm3+進行包覆,得到核殼結構的YPO4∶Sm3+@YPO4納米熒光粉。然后以PEG為有機表面改性劑,采用微波法對其表面改性,得到雙層核殼結構的熒光納米材料YPO4∶Sm3+@YPO4@PEG。結果表明:包覆前后的產物均為四方晶系結構,產物形貌均為納米球,包覆后產物大小分布均勻且顆粒分散性較好,表面光滑、圓潤。包覆層的厚度為10~20 nm。包覆后產物熒光光譜的激發和發射峰的位置未發生移動,但熒光強度與包覆前相比顯著提升,其中YPO4∶Sm3+與包覆劑YPO4物質的量之比為6∶1時所得產物的熒光強度最大,是YPO4∶Sm3+熒光強度的4倍多。PEG包覆量為0.2 g時,產物的熒光強度增強了5倍多。包覆前后色坐標略有變化,顏色為橙紅色。該雙層核殼結構的納米熒光粉YPO4∶Sm3+@YPO4@PEG不僅具有親水性和生物相容性,而且增強了YPO4∶Sm3+的發光性能,在生物醫藥領域中有潛在的應用價值。

- 無機化學學報的其它文章
- La-Doped BaSnO3/Multi-walled Carbon Nanotube Modified Separator:Synthesis and Application in Lithium-Sulfur Battery
- Co(Ⅱ)/Ni(Ⅱ) Coordination Polymer of Isomeric Terphenyl-2,2″,4,4″-tetracarboxylic Acids with a Single Water Bridge:Syntheses,Structures,and Magnetic Properties
- Micromotors Based on Ni-Mn Binary Oxide and Its Application for Effective Dye Adsorption
- Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol by Mg-Doped Ceria Monolithic Catalyst
- Hydrogen Storage Capabilities of the Low-Lying Ca2B4Clusters
- 盤狀鏑簇合物的合成及緩慢磁弛豫