羚羊角水解液的氨基酸薄層鑒別與含量測定
2022-07-18 08:07:30劉妍劉靜陳揚趙苑伶張雪陳林珍趙崇軍馬志強
環球中醫藥 2022年7期
劉妍 劉靜 陳揚 趙苑伶 張雪 陳林珍 趙崇軍 馬志強
羚羊角為牛科動物賽加羚羊SaigatataricaLinnaeus的角,是傳統名貴中藥材之一,始載于《神農本草經》,在我國已有2000多年的藥用歷史。羚羊角性寒,味咸,歸肝、心經,具有平肝息風、清肝明目、散血解毒的功效,主治肝風內動、驚癇抽搐、妊娠子癇、高熱痙厥、癲癇發狂、頭痛眩暈、目赤翳障、溫毒發斑、癰腫瘡毒[1]。羚羊角顆粒是由羚羊角水解液經加工而成的現代藥物制劑,氨基酸是羚羊角的主要成分之一[2-3]。目前筆者未見國內文獻用微乳液來分離鑒別動物中的氨基酸。微乳薄層色譜(microemulsion thin layer chromatography,METLC)的展開劑是微乳液,因其具有較大的增溶量和超低界面張力,能讓難以分離的復雜體系和難溶化合物得以分離[4-6],近些年來已經成功運用于生物堿[7]、黃酮[8-11]、糖類[12]、氨基酸[13]等多種化合物的分離鑒別中。為了更好地對羚羊角水解液中的氨基酸進行分析,本實驗采用十二烷基硫酸鈉(sodium dodecyl sulfate,SDS)—正丁醇—正己烷—水按質量比(1.5∶7.0∶1.0∶1.7)為展開劑對羚羊角水解液中的氨基酸進行定性鑒別,用異硫氰酸苯酯柱前衍生法對水解液中16種游離氨基酸進行含量測定,進一步豐富了微乳液應用于氨基酸薄層鑒別研究的方法,也為羚羊角藥材的質量控制提供研究基礎。
1 儀器與材料
1.1 實驗儀器
十萬分之一分析天平(Mettler Toledo公司),循環水式多用真空泵(鄭州長城科工貿有限公司),N-1100型旋轉蒸發儀,水浴鍋(上海愛朗儀器有限公司),Sorvall ST 8R臺式高速冷凍離心機(美國Thermo Fisher Scientific公司),硅膠G板(100 mm×100 mm,青島海洋化工廠分廠,批號:20181205、20190322、20190802),硅膠G板(企業A,批號:20200410),1 μL、2 μL定量點樣毛細管(潤澤康),雙槽P-1型薄層色譜展開缸(100 mm×100 mm),DHG-9023A型電熱恒溫鼓風干燥箱(上海精宏實驗設備有限公司),高效液相色譜儀(Waters 2695)包括2695四元梯度泵,自動進樣器,2998 PDA檢測器,在線脫氣機,柱溫箱,Empower色譜工作站。
1.2 實驗藥物
羚羊角對照藥材,中國食品藥品檢定研究院,批號:PS210423-04;羚羊角粉(過200目篩),企業B,批號:20201011,由北京中醫藥大學楊瑤珺教授鑒定為羚羊角粉;羚羊角水解液,實驗室自制。
1.3 實驗試劑
16種氨基酸對照品購自上海源葉生物科技有限公司,天冬氨酸Asp(批號:S24A8I34463,純度≥99%)、谷氨酸Glu(批號:S27M6G1,純度≥99%)、絲氨酸Ser(批號:S04J9I51507,純度≥98%)、甘氨酸Gly(批號:SM0315GA14,純度≥99%)、組氨酸His(批號:Z19A9H59384,純度≥98%)、精氨酸Arg(批號:MKBD3032V,純度≥98%)、蘇氨酸Thr(批號:S01F4G1,純度≥98%)、丙氨酸Ala(批號:S20A6G17672,純度≥98%)、脯氨酸Pro(批號:S30J6G1,純度≥99%)、酪氨酸Tyr(批號:SM0503GE13,純度≥99%)、纈氨酸Val(批號:S06D8I49809,純度≥98%)、甲硫氨酸Met(批號:SLBK1770V,純度≥98%)、異亮氨酸Ile(批號:SM0503GD13,純度≥98%)、亮氨酸Leu(批號:S10D9I76995,純度≥98%)、苯丙氨酸Phe(批號:H20N8H48638,純度≥98%)、賴氨酸Lys(批號:S26A8I34751,純度≥98%)。SDS,購自湖南爾康制藥股份有限公司,批號:101420181101;正丁醇、無水乙酸鈉、濃鹽酸、乙酸(分析純),均購自北京化工廠,批號依次為:20170816、20180605、20180712、20170215;乙腈、正己烷(色譜純),購自費希爾控制設備國際有限公司,批號依次為:190263、192565;三乙胺(分析純),購自上海麥克林生化科技有限公司,批號:C10487468;異硫氰酸苯酯購自博納艾杰爾,批號:BCBB1912V;茚三酮(分析純)、乙醇(分析純),均購自于福晨(天津)化學試劑有限公司,批號依次為:20190402、20210108。
2 方法與結果
2.1 羚羊角水解液的薄層色譜分析
2.1.1 對照品溶液的制備 精密稱取L-天冬氨酸、L-谷氨酸、L-纈氨酸、L-亮氨酸、L-賴氨酸對照品適量,分別加入50%甲醇、50%甲醇、70%甲醇、70%甲醇、90%甲醇制成0.3 mg/mL的對照品溶液。
2.1.2 對照藥材、供試品溶液的制備 精密稱取羚羊角對照藥材及羚羊角粉(過200目篩)1.0 g置于100 mL圓底燒瓶中,第一次加入40 mL水,回流提取3小時,抽濾。第二次加入40 mL水,回流提取2小時,抽濾,合并兩次濾液。剩余藥渣中加入16 mL 2 mol/L硫酸溶液回流水解6小時,抽濾后,濾液中加入調成糊狀的氫氧化鈣,調pH值為4,沉淀24小時,抽濾得其濾液,將之與水提取濾液合并濃縮后進行60%醇沉,在4℃下沉淀24小時。抽濾,濃縮,用水定容至10 mL。再以5 000 r/分鐘的轉速離心20分鐘得其上清液即為羚羊角水解液的供試品溶液。
2.1.3 微乳液展開劑的配制 將SDS、正丁醇、正己烷、水按質量比(1.5∶7.0∶1.0∶1.7)混勻即得含水量15%的微乳液[14-15]。
2.1.4 羚羊角水解液的薄層鑒別 參考2020版《中華人民共和國藥典(四部)》通則0502[16],吸取天冬氨酸、谷氨酸、纈氨酸、亮氨酸、賴氨酸對照品溶液各2 μL,吸取供試品溶液及對照藥材溶液0.5 μL分別點于事先在110℃活化半小時的同一硅膠G薄層板上,以含水量為15%的微乳液上行展開,展距為8 cm時將薄層板取出,晾干,噴以2%茚三酮乙醇溶液,在105℃烘至斑點顯色清晰。
自制8批羚羊角水解液的薄層圖譜見圖1。羚羊角水解液除與對照品色譜相應位置上顯集中且圓的斑點外,與對照藥材所示色譜條帶一致,分離效果佳,天冬氨酸、谷氨酸、纈氨酸、亮氨酸、賴氨酸的Rf值依次為0.20、0.14、0.50、0.61、0.33。
2.2 羚羊角水解液的薄層耐用性考察
2.2.1 不同薄層板的分離效果考察 取供試品溶液、對照品溶液按2.1.4項下方法點于不同廠家薄層板(企業A、青島海洋化工廠分廠)及同一廠家不同批號薄層板(青島海洋化工廠分廠,批號:20181205、20190322、20190802),見圖2。青島海洋化工廠分廠所制薄板分離效果要優于企業A。
2.2.2 不同溫度的分離效果考察 取供試品溶液、對照品溶液按2.1.4項下方法點于同一批號薄層板,分別置于4℃與30℃的環境下展開,見圖3。羚羊角水解液的色譜圖在4℃與30℃下大致一致。

注:1 天冬氨酸;2 谷氨酸;3 纈氨酸;4 亮氨酸;5 賴氨酸;6~8 自制3批羚羊角水解液。A:企業A;B、C、D:青島海洋化工廠分廠,B:批號為20181205,C:批號為20190322,D:批號為20190802。

注:1 天冬氨酸;2 谷氨酸;3 纈氨酸;4 亮氨酸;5 賴氨酸;6~8 自制3批羚羊角水解液。A:溫度4℃;B:溫度30℃。
2.2.3 不同濕度的分離效果考察 取供試品溶液、對照品溶液按2.1.4項下方法點于同一批號薄層板,分別置于相對濕度為32%、58%、88%的環境下展開,結果見圖4。羚羊角水解液的色譜圖在相對濕度32%、58%、88%環境下基本相同。

注:1 天冬氨酸;2 谷氨酸;3 纈氨酸;4 亮氨酸;5 賴氨酸;6~8 自制3批羚羊角水解液。A:相對濕度32%;B:相對濕度58%;C:相對濕度88%。
2.2.4 不同點樣量的分離效果考察 取供試品溶液、對照品溶液按2.1.4項下方法點于同一薄層板上,供試品點樣量由左往右依次為0.5 μL、1 μL、1.5 μL,結果見圖5。羚羊角水解液最佳點樣量為0.5 μL,其次為1 μL,當點樣量為1.5 μL時拖尾明顯。

注:1 天冬氨酸;2 谷氨酸;3 纈氨酸;4 亮氨酸;5 賴氨酸;6~8點樣量依次為0.5 μL、1 μL、1.5 μL。
2.2.5 不同含水量微乳液的分離效果考察 取供試品溶液、對照品溶液按2.1.4項下方法點于同一批號薄層板上,在固定SDS、正丁醇、正己烷質量比為1.5∶7.0∶1.0的配比下,分別用含水量為10%、13%、15%、17%、20%的微乳液展開,結果見圖6。各含水量圖譜基本一致。
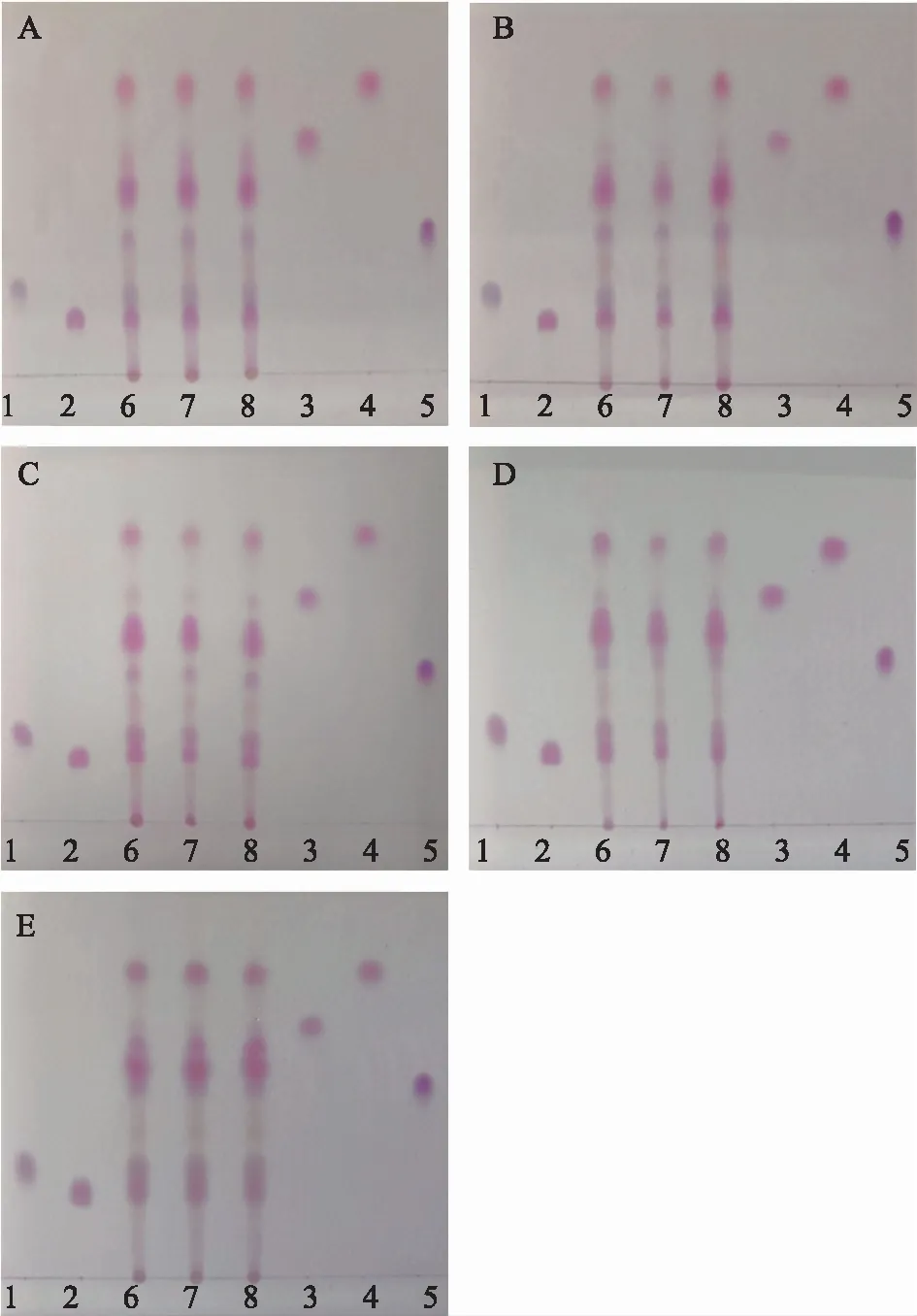
注:1 天冬氨酸;2 谷氨酸;3 纈氨酸;4 亮氨酸;5 賴氨酸;6~8 自制3批羚羊角水解液。A:含水量10%;B:含水量13%;C:含水量15%;D:含水量17%;E:含水量20%。
2.3 羚羊角水解液中游離氨基酸的分析
2.3.1 色譜分析條件 Venusil AA氨基酸分析專用柱(博納艾杰爾,4.6×250 mm,5μm),柱溫40℃,樣品室溫度為4℃,流速1.0 mL/分,檢測波長254 nm,一次進樣量10 μL。流動相A:0.1 mol/L乙酸鈉(pH 6.5)—乙腈(93∶7),流動相B為80%乙腈,梯度洗脫:0分鐘:0% B;14分鐘:9.5% B;22分鐘:19.5% B;37分鐘:30.6% B;41分鐘:31.6% B;42分鐘:34.0% B。
2.3.2 衍生化溶液的制備 三乙胺乙腈溶液:將1.4 mL的三乙胺加入到8.6 mL的乙腈中混勻即得;異硫氰酸苯酯乙腈溶液:將25 μL的異硫氰酸苯酯加入到2 mL乙腈中混勻即可。
2.3.3 混合對照品溶液的配制 分別稱取天冬氨酸 33.28 mg、谷氨酸36.78 mg、絲氨酸 26.31 mg、甘氨酸18.70 mg、組氨酸 38.81 mg、精氨酸 43.56 mg、蘇氨酸29.76 mg、丙氨酸 22.23 mg、脯氨酸28.82 mg、酪氨酸 45.26 mg、纈氨酸 29.24 mg、甲硫氨酸 37.33 mg、異亮氨酸32.75 mg、亮氨酸 32.86 mg、苯丙氨酸 41.29 mg、賴氨酸 36.50 mg,加入0.1 mol/L鹽酸溶液超聲溶解并定容至100 mL。
2.3.4 供試品溶液的制備 稱取5 g羚羊角粉末(過200目篩),第一次加入200 mL水回流提取3小時,抽濾;第二次加入200 mL水回流提取2小時,抽濾。濾渣用80 mL 2 mol/L的硫酸溶液回流水解6小時,抽濾后用調成糊狀的氫氧化鈣調至pH值為4,沉淀24小時,濾過后將酸水解濾液與水提取濾液合并濃縮進行60%醇沉,4℃沉淀24小時。濾過后回收乙醇定容至20 mL,即得羚羊角水解液,5000 r/min離心20分鐘。精密吸取離心后的上清液2 mL,加入6 mL乙腈,搖勻過濾,用乙腈—水溶液(3∶1)清洗沉淀,旋轉蒸發濃縮至干,用0.1 mol/L鹽酸定容至5 mL。
2.3.5 標準品與供試品的衍生 精密吸取200 μL 0.1 mol/L鹽酸溶液、氨基酸混合對照品溶液及供試品溶液于1.5 mL離心管中,加入100 μL三乙胺乙腈溶液和100 μL異硫氰酸苯酯乙腈溶液,混勻,于室溫下放置1小時。接著加入400 μL正己烷,振搖后放置10分鐘,取下層溶液,過0.22 μm濾膜,取濾液200 μL,加800 μL純凈水稀釋,搖勻制得空白衍生化溶液、氨基酸對照衍生化溶液和供試品衍生化溶液。圖譜如圖7所示。
2.4 方法學考察
2.4.1 線性關系考察 精密稱取對照品天冬氨酸、谷氨酸、絲氨酸、組氨酸、精氨酸、蘇氨酸、丙氨酸、甲硫氨酸適量,用0.1 mol/L鹽酸溶液逐級稀釋,將對照品溶液配制成0.0125 μmol/mL、0.0625 μmol/mL、0.125 μmol/mL、0.25 μmol/mL、0.625 μmol/mL、1.25 μmol/mL、2.5 μmol/mL;精密稱取對照品甘氨酸、脯氨酸、酪氨酸、纈氨酸、異亮氨酸、亮氨酸、苯丙氨酸、賴氨酸適量,用0.1 mol/L鹽酸溶液逐級稀釋,將對照品溶液配制成0.0069 μmol/mL、0.0625 μmol/mL、0.125 μmol/mL、0.25 μmol/mL、0.625 μmol/mL、1.25 μmol/mL、2.5 μmol/mL。按2.3.5項下分別衍生化,再按2.3.1項下色譜條件進樣。以各氨基酸的濃度(μmol/mL)為橫坐標,相應色譜峰峰面積為縱坐標制作線性回歸方程。回歸方程具體見表1。
2.4.2 檢測限和定量限 將氨基酸對照品溶液依次稀釋衍生進樣,分別以16種氨基酸的信噪比為3∶1和10∶1時確定檢測限和定量限,具體見表1。
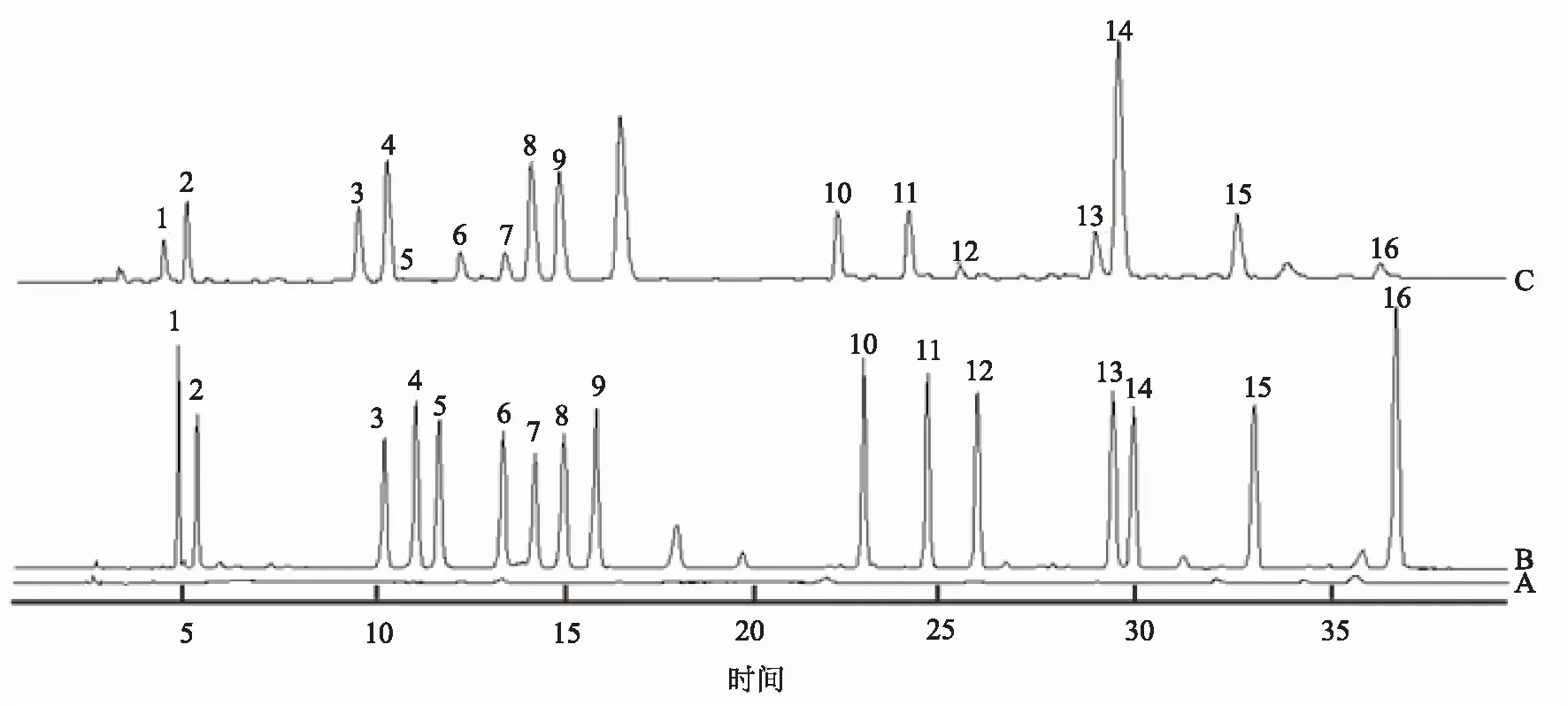
注:1 天冬氨酸;2 谷氨酸;3 絲氨酸;4 甘氨酸;5 組氨酸;6 精氨酸;7 蘇氨酸;8 丙氨酸;9 脯氨酸;10 酪氨酸;11 纈氨酸;12 甲硫氨酸;13 異亮氨酸;14 亮氨酸;15 苯丙氨酸;16 賴氨酸。A 空白衍生化溶液;B 氨基酸對照衍生化溶液;C 供試品衍生化溶液。
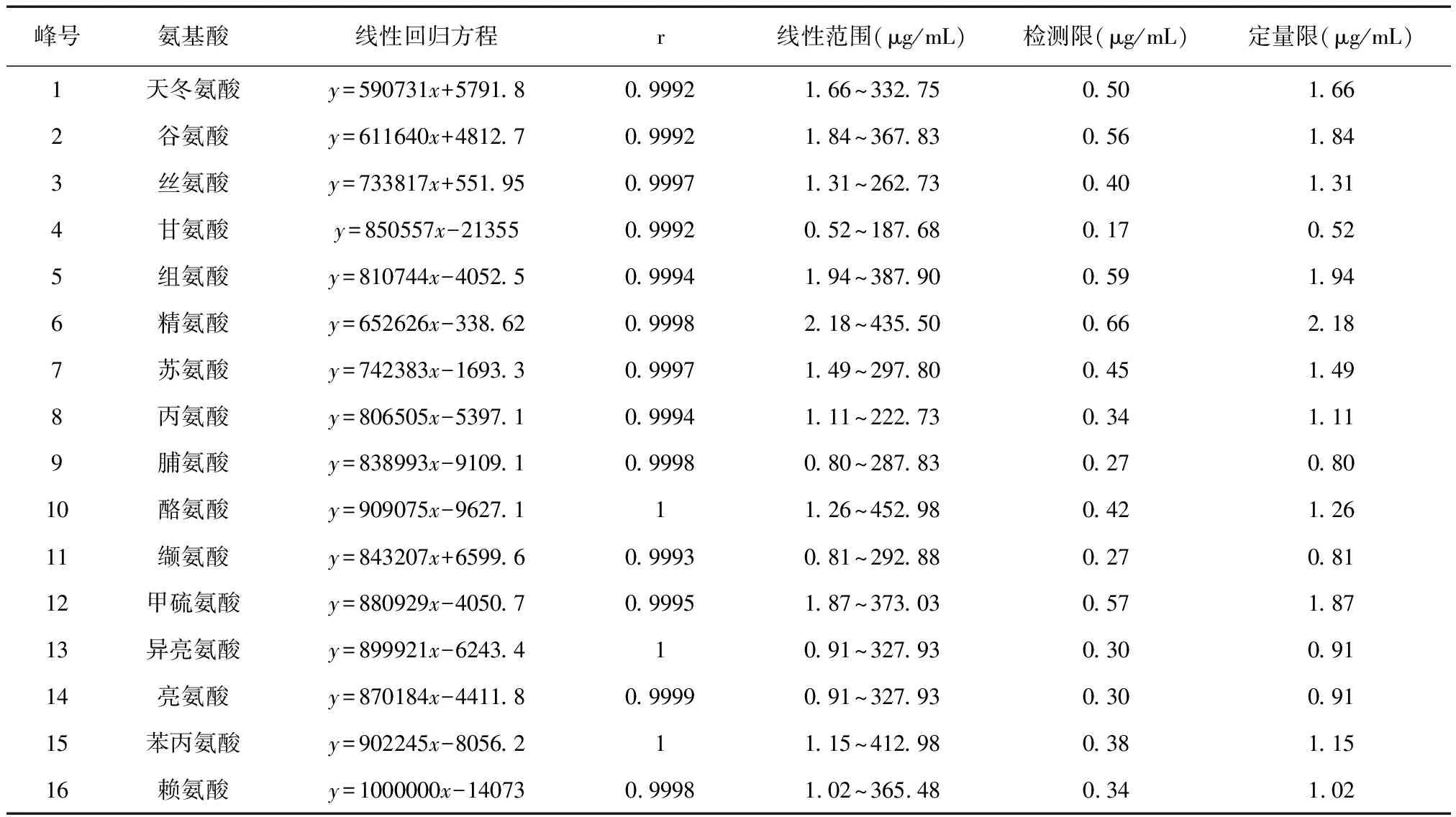
表1 16種氨基酸的相關參數
2.4.3 精密度試驗 將同一瓶供試品溶液,按2.3.1項下色譜條件連續進樣6次,記錄各氨基酸的峰面積。羚羊角水解液中16種游離氨基酸峰面積的相對標準偏差(relative standard deviation,RSD)為0.75%~2.28%,表明儀器精密度良好。
2.4.4 穩定性試驗 將供試品衍生化溶液于制備后0小時、4小時、8小時、12小時、16小時、24小時進樣分析,結果顯示羚羊角水解液中16種游離氨基酸峰面積的RSD為0.72%~2.72%,這表明供試品衍生化溶液在4℃、24小時內穩定。
2.4.5 重復性試驗 取同一批次水解液平行制樣6份,測定氨基酸的峰面積,羚羊角水解液中16種游離氨基酸峰面積的RSD為0.81%~2.59%,說明此法重復性良好。
2.4.6 回收率試驗 取6份同一批已測定含量的羚羊角水解液的上清液1 mL,加入3 mL乙腈,搖勻過濾,用乙腈∶水溶液(3∶1)清洗沉淀,旋轉蒸發濃縮至干,加入與之含量相近的氨基酸混合對照品,用0.1 mol/L鹽酸定容至5 mL。羚羊角水解液中16種游離氨基酸的平均回收率(n=6)在96.70%~103.02%之間,RSD均<3%,符合要求。
2.4.7 樣品含量測定 將自制的8批羚羊角水解液按“2.3.4、2.3.5”項下制樣衍生,隨后按“2.3.1”項下色譜條件進樣,記錄各氨基酸的峰面積,代入相應氨基酸的線性方程計算各氨基酸含量,結果以羚羊角藥材與水解液體積比例為1∶1時表示,具體見表2。
3 討論
用含水量15%的微乳液即SDS—正丁醇—正己烷—水按質量比(1.5∶7.0∶1.0∶1.7)進行配比用于薄層色譜展開時,用恰當適宜的點樣量,即便溫度、相對濕度發生變化,羚羊角水解液中的天冬氨酸、谷氨酸、纈氨酸、亮氨酸、賴氨酸這5種氨基酸均能實現良好分離,說明所建立的微乳薄層方法可以應對全國各地不同氣候對結果的影響。羚羊角水解液中游離氨基酸含纈氨酸、異亮氨酸、亮氨酸、苯丙氨酸、甲硫氨酸、蘇氨酸、賴氨酸這7種人體必需氨基酸,也含有對嬰兒而言所必需的組氨酸,此外亦有天冬氨酸、谷氨酸、絲氨酸、甘氨酸、精氨酸、丙氨酸、脯氨酸、酪氨酸。曾對液相流速(0.6 mL/分鐘、0.8 mL/分鐘、1.0 mL/分鐘)及溫度(30℃、35℃、40℃)進行考察,發現流速為0.6 mL/分鐘時,4號峰甘氨酸與5號峰組氨酸不能完全分離,而流速0.8 mL/分鐘能使甘氨酸與組氨酸的色譜峰完全分開但較流速為1.0 mL/分鐘耗費的時間長。固定流速為1.0 mL/分鐘考察溫度,溫度為30℃時,發現8號峰丙氨酸與9號峰脯氨酸無法分離,35℃時這兩個色譜峰可以分離,由于在其他條件一定時,柱溫上升會使容量因子變小,使色譜峰流出加快,綜合考慮在保證分離度的前提下,為省時選擇流速為1.0 mL/分鐘、柱溫為40℃。本次實驗沒有考察微乳液中表面活性劑、助表面活性劑、油相的種類與占比對羚羊角水解液的薄層色譜圖的影響,期待后續研究能完善這部分。
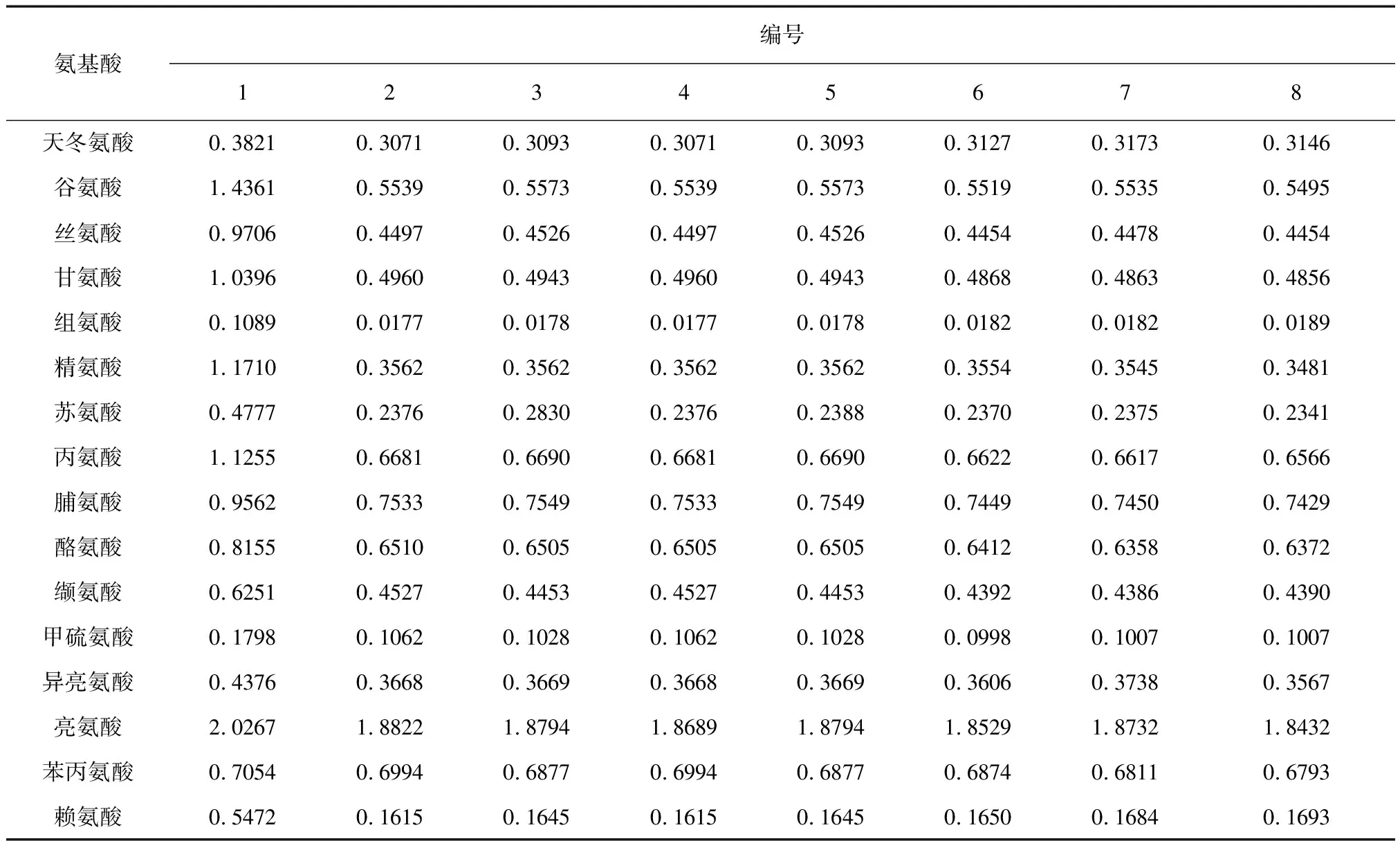
表2 羚羊角水解液中游離氨基酸的含量(mg/mL)
筆者也曾用經典的正丁醇—冰醋酸—水即“BAW系統”進行展開,無論怎樣調試比例亦或是增加甲醇、丙酮、二氯甲烷使展開系統變成五元乃至六元皆無法讓羚羊角水解液中的5種氨基酸獲得良好分離,而采用W/O型微乳液作為展開劑卻能達到分離的目的,這可能是因為微乳色譜中,溶質在固定相、油或水連續相及內核和介面膜等數相之間進行分配,層析過程是集吸附、分配、靜電、疏水、立體、萃取與反萃取等多效應于一體,從而使各物質遷移速率不同而使Rf值有差異[17-20]。與傳統薄層色譜相較,微乳薄層色譜有分離效果顯著提高、分離的成分增多、斑點圓而集中、重現性好、點樣量少等優勢[21-22],在中藥有效成分的分析應用中也越來越廣泛。筆者將微乳薄層色譜應用于羚羊角水解液的氨基酸分離鑒別中,進一步豐富了W/O型微乳用于氨基酸薄層分離分析的研究,亦為羚羊角藥材的質量控制提供科學依據。
