人類皰疹病毒逃逸宿主抗病毒固有免疫應答機制的研究進展
2022-08-09 07:22:50牛宇輝李向茸馮若飛
中國人獸共患病學報 2022年1期
關鍵詞:信號
牛宇輝,李向茸,馮若飛
皰疹病毒(Herpesvirus)是一類有囊膜的雙鏈DNA病毒,其基因組大小約為125~235 kb,病毒顆粒為對稱的正二十面體結構[1]。根據該科病毒生物學特性和基因組結構的不同可將其劃分為α、β和γ 3個亞科,在這3個亞科中共發現了9種可引起人類疾病的病毒被統稱為人類皰疹病毒(Human herpesvirus, HHV)[1],其中屬于α皰疹病毒亞科的病毒有單純皰疹病毒I型(Herpes simplex virus-1, HSV-1)、單純皰疹病毒II型(Herpes simplex virus-2, HSV-2)、水痘-帶狀皰疹病毒(Varicella-zoster virus, VZV);β皰疹病毒亞科的人巨細胞病毒(Human cytomegalovirus, HCMV)、人類皰疹病毒6A型(Human herpesvirus-6A, HHV-6A)、人類皰疹病毒6B型(Human herpesvirus-6B, HHV-6B)、人類皰疹病毒7型(Human herpesvirus-7, HHV-7)及γ皰疹病毒亞科的愛潑斯坦-巴爾病毒(Epstein-barr virus, EBV)和卡波西氏肉瘤相關皰疹病毒(Kaposi’s sarcoma-associated herpesvirus, KSHV)。在人類皰疹病毒與宿主細胞長期的共進化過程中,病毒進化出多種策略以逃避或干擾宿主細胞介導的免疫應答反應。研究發現HHV病毒蛋白及其miRNA可通過干擾固有免疫信號通路、細胞自噬和內質網應激等方式破壞機體免疫應答以達到促進自身增殖的目的[2-3]。基于此,本文概述近年來人類皰疹病毒逃避宿主固有免疫應答的具體機制,以期能夠對人類皰疹病毒相關抗病毒藥物及疫苗研發工作提供全面而系統的研究思路。
1 人類皰疹病毒對宿主固有免疫應答的激活作用
固有免疫系統是抗體抵御病毒感染并啟動宿主細胞免疫級聯反應的首道宿主防線,其產生的細胞因子和炎性介質是抵抗病毒等外源性微生物的主要武器[4]。當外源微生物或危險信號刺激機體后,宿主細胞編碼的模式識別受體(Pattern recognition receptors, PRRs)識別病原相關分子模式(Pathogen-associated molecular patterns, PAMPs)或損傷相關分子模式(Damage associated molecular patterns, DAMPs),進而啟動宿主細胞的固有免疫反應[4]。一般情況下,病毒的蛋白成分首先會被細胞表面的Toll樣受體(Toll-like receptors, TLRs)所識別,而入侵到細胞質中的病毒成分則主要被細胞內的C型植物血凝素受體(C-type lectin receptors, CLRs)、核苷酸結合寡聚化結構域NOD樣受體(Nucleotide-binding oligomerization domain like receptors, NLRs)、視黃酸誘導型基因樣受體(Retinoic acid inducible gene-I-like receptors, RLRs)以及DNA識別受體等PRRs所識別,進而激活機體的抗病毒防御機制,降低病毒的感染能力[4]。在病毒感染的不同階段所啟動的免疫反應不盡相同,不同的病毒所啟動的固有免疫機制也不相同。已有文獻報道了人類皰疹病毒可被TLR1、TLR2及TLR6識別,之后它們在腫瘤壞死因子受體相關因子6(TNF receptor-associated factor 6, TRAF6)和髓樣分化因子88(Myeloid differentiation factor 88, MyD88)存在的條件下激活核轉錄因子(Nuclear factor κB, NF-κB)介導的炎癥因子和細胞因子的分泌,抑制病毒在宿主細胞中的增殖[5]。除TLRs和RLRs外,還有幾種新發現的DNA識別受體,如黑色素瘤缺乏因子2(Absent in melanoma 2,AIM2)、DEAD-box helicase 41(DDX41)、DNA依賴的干擾素調節因子激活物(DNA-dependent activator of IFN-regulatory factors, DAI)、cGAMP合成酶(cyclic Guanosine monophosphate-adenosine monophosphate synthase, cGAS)和γ干擾素誘導蛋白16(IFN-γ-inducible protein 16, IFI16)也可與人類皰疹病毒的相應配體結合,通過MyD88、線粒體抗病毒信號接頭蛋白(Mitochondrial antiviral signaling protein, MAVS)和干擾素刺激基因(Stimulator of interferon genes, STING)等向下游傳遞活化信號,促使干擾素調節因子3/7(Interferon regulatory factors 3/7, IRF3/7)和核轉錄因子NF-κB的激活,進而誘導干擾素和炎性細胞因子的表達,激活宿主細胞的固有免疫應答級聯反應[6]。
在病毒感染早期,皰疹病毒衣殼蛋白首先在細胞質中經過泛素化后被蛋白酶體途徑所降解,隨后將基因組DNA釋放至細胞質,并被DNA識別受體識別。在皰疹病毒大量感染細胞時,在胞質中釋放的病毒dsDNA或異常定位的細胞DNA被cGAS所識別,并催化第二信使環鳥甘酸-腺苷酸(cyclic Guanosine monophosphate-adenosine monophosphate, cGAMP)與內質網(Endoplasmic reticulum, ER)中的STING相結合,使其二聚化激活后從ER轉移至高爾基體,隨后招募TKB1和IκB激酶,進而磷酸化IRF3和IκBα,激活NF-κB和IFN-I的轉錄[7-9]。在皰疹病毒感染時,IFI16也可以移位到細胞質并誘導STING介導的信號通路的激活[6]。人類皰疹病毒不僅具有強大的編碼能力和建立終身感染的能力,其在病毒感染宿主細胞過程中,還可通過“劫持”多種細胞進程、主動調控或改變細胞內環境,為自身的復制創造有利條件。例如,HHV必須借用宿主細胞的蛋白合成系統合成自身蛋白,用于組裝子代病毒粒子;細胞出于自我保護的目的會啟動未折疊蛋白反應(Unfolded protein response, UPR)介導的蛋白降解、細胞凋亡、炎癥反應、自噬等抑制HHV病毒蛋白的合成、降解病毒蛋白或誘導伴侶分子表達,從而誘導內質網應激[10-12]。由此可見,宿主細胞固有免疫系統可通過多種機制來阻止HHV感染。與此同時,HHV也進化出了各種各樣逃避宿主免疫反應的策略。因此,掌握HHV與宿主固有免疫應答之間的關系將為探索新型抗人類皰疹病毒的藥物作用靶點提供研究思路。
2 人類皰疹病毒對宿主固有免疫應答的逃逸機制
在宿主細胞與病毒的長期共進化過程中,病毒的結構蛋白和非結構蛋白也進化出了多樣的逃避宿主固有免疫應答反應的策略,為自身的增殖創造機會。皰疹病毒幾乎能在所有的動物中建立潛伏感染,且潛伏感染期間病毒保持休眠狀態,宿主不表現出任何臨床癥狀,而在機體受到一些刺激引起應激反應后,潛伏的病毒被重新激活,誘導機體出現復發性感染[13]。大部分HHV是通過抑制cGAS-STING途徑介導的IFN-I信號通路和核轉錄因子NF-κB中關鍵接頭蛋白的激活,進而抑制干擾素及炎性細胞因子的產生,最終促進病毒的增殖,以下分別闡述各種人類皰疹病毒逃逸宿主固有免疫應答反應的機制,具體見圖1。
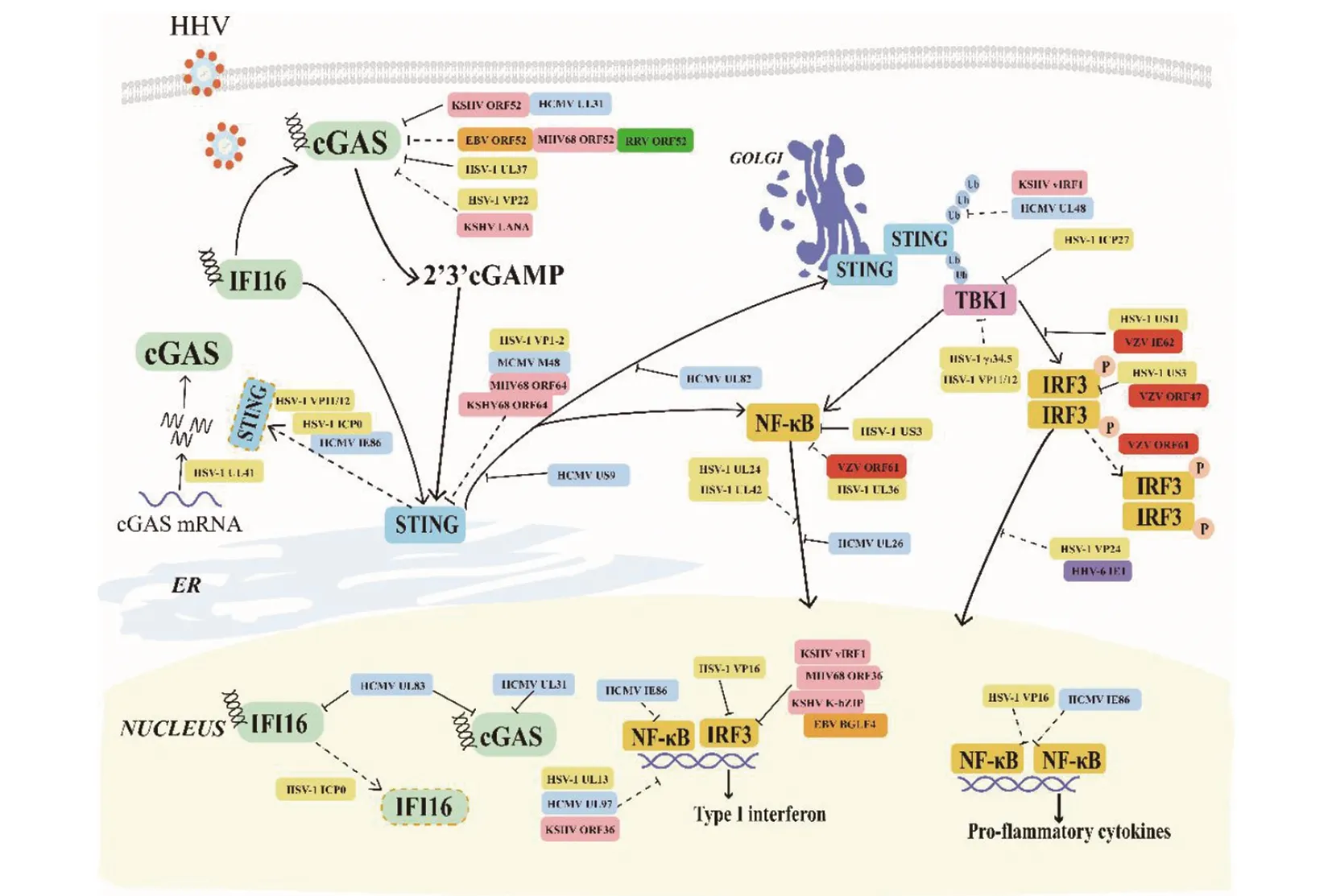
cGAS:環GAMP合成酶;STING:干擾素刺激基因;IFI16:γ-干擾素誘導蛋白16;TBK1:Tank結合激酶1;IRF3:干擾素調節因子3;NF-κB:核轉錄因子;“實線箭頭”代表一種蛋白可直接促進另一種蛋白的表達;“平箭頭”代表發揮抑制作用;“虛線箭頭”代表一種蛋白可間接影響另一種蛋白的表達。
2.1 HSV逃避宿主固有免疫應答反應的方式 HSV有HSV-1型和HSV-2型。HSV-1感染是造成原發性和復發性水皰性皮疹的主要原因之一,感染部位集中在皮膚黏膜和腦部[11],而HSV-2感染多發生在陰莖、陰道、子宮頸或臀部等生殖器官或鄰近部位。HSV通過拮抗干擾素信號通路相關途徑關鍵靶蛋白和抑制宿主細胞凋亡來干擾宿主抗病毒機制,促進病毒在機體內的增殖。在病毒感染的早期,HSV可被細胞表面受體TLRs識別,HSV進入細胞質后可被DNA和RNA傳感器識別。研究發現,多個HSV蛋白參與逃避宿主RLRs信號通路,如HSV-2 VHS通過選擇性下調TLR-2、TLR-3、RIG-I和MDA5來抑制IFN信號通路[14];HSV-1 US11通過與RIG-I和MDA5的C末端RNA結合結構域競爭性結合RNA,阻礙下游信號傳遞從而拮抗IFN-I信號通路[15];US11還可與Hsp90形成復合物替代TBK1,從而干擾HSV-1感染后IRF3與TBK1的相互作用以及阻礙IRF3磷酸化[16]。HSV-2 US1可與IRF3的DNA結合域相互作用,進而抑制IRF3與IFN-β啟動子的結合,從而抑制IFN-β的產生[17];HSV-1 UL37通過與RIG-Ⅰ及cGAS結合并介導其脫酰胺化,使其功能受損,從而阻斷其信號傳遞[18];HSV-1 VP16可與IRF3的轉錄共激活因子CREB結合蛋白(CREB binding protein,CBP)相結合,抑制了CBP向IRF3的募集,進而抑制IRF3-CBP復合體與IFN-β啟動子的結合,從而抑制IFN-I的產生[15];HSV-1 UL42通過與NF-κB的亞基p65/p50的Rel同源結構域(Rel homology domain, RHD)結合并劫持p65和p50,阻礙其入核,從而抑制NF-κB的激活[19];HSV-1 US3結合IRF3和NF-κB的p65亞基,并依賴其激酶活性異常磷酸化IRF3和p65,抑制它們的二聚化及其入核,從而拮抗IFN-I信號通路[20];HSV-1 ICP0可以分別與NF-κB的p65和p50相結合以抑制NF-κB的激活,但具體機制不同,HSV-1 ICP0與p65的RHD結構域結合,抑制腫瘤壞死因子α(Tumor necrosis factor α, TNFα)誘導的p65的入核,抑制NF-κB的轉錄;HSV-1 ICP0與p50結合依賴其E3泛素連接酶活性降解p50,抑制NF-κB的轉錄[21]。HSV基因組DNA進入宿主細胞質后,cGAS-STING信號通路便發揮主要的抗病毒作用,而HSV也通過自己的方式拮抗宿主抗病毒反應。例如,HSV-1 VP22可通過與cGAS結合并抑制其酶活性,減少cGAMP的生成,從而抑制STING激活,最終抑制IFN-I的產生[22];HSV-1 ICP0還可依賴其E3泛素酶活性降解IFI16,從而阻斷該傳感器下游的信號傳導;HSV-1 VP24通過干擾TBK1與IRF3的相互作用抑制STING下游的信號傳導[23];HSV-1 UL36通過去泛素化IκBα抑制其降解,進而阻斷cGAS和STING對NF-κB的激活,最終抑制IFN-I的產生[15];HSV-1 UL24通過與p65和 p50相互作用,阻礙它們的入核,抑制cGAS和STING對NF-κB的激活,減少IFN-I的產生[24];HSV-1 UL41依賴其核酸內切酶活性降解IFI16和cGAS的mRNA,干擾宿主對HSV-1的識別,從而拮抗IFN-I信號通路[25];HSV-1 VP11/12(由UL46編碼)通過介導STING的降解來阻斷STING介導的信號傳遞[26];HSV-1 ICP27通過靶向TBK1介導的IRF3磷酸化負調控IFN-I反應[27];此外,HSV還可抑制JAK/STAT信號通路,比如HSV-1 UL41依賴其激酶活性降解鋅指抗病毒蛋白(Zinc-finger antiviral protein, ZAP)、三角形四肽重復干擾素誘導蛋白3(Interferon-induced protein with tetratricopeptide repeats 3, IFIT3)和抗病毒蛋白viperin的mRNA,下調它們的表達,逃避干擾素刺激基因(IFN-stimulated genes, ISGs)的抗病毒作用[15];HSV-1 UL36通過特異性靶向結合Ⅰ型干擾素受體2(Type I IFN receptor 1, IFNAR2),抑制IFNAR2與JAK1結合,從而拮抗IFN-I信號通路[15]。HSV miRNA也在逃避固有免疫反應過程中發揮著作用。HSV-1 miR-H1可抑制細胞吞噬作用,還可下調多種細胞因子如TNFα、IL-1β和IL-10的表達水平[28]。
2.2 EBV逃避宿主固有免疫應答反應的方式 EBV是一種常見的致瘤性病毒,最初發現于Burkitt淋巴瘤,與惡性腫瘤如彌漫性大B細胞淋巴瘤、霍奇金淋巴瘤和惡心淋巴增殖性疾病等有關[11]。EBV病毒蛋白及其miRNA在逃避機體免疫、促進病毒感染過程中發揮著重要作用。在感染早期EBV可使TLRs受體的表達降低,如EBV BGLF5蛋白可以選擇性下調TLR2和TLR9的表達[29];EBV BGLF4還可使NF-κB的共激活因子泛表達轉錄子(Ubiquitously expressed transcript, UXT)磷酸化來抑制NF-κB的活性[29];EBV 去泛素化酶BPLF1阻礙了TLRs信號中間產物的泛素化,抑制NF-κB的激活[29]。還有許多EBV病毒蛋白在干擾宿主細胞IFN-I及JAK/STAT通路信號傳導過程中發揮著重要作用,如EBV BGLF4可使IRF3在S123、S173和T180位點去磷酸化,進而抑制干擾素及其下游因子的表達[3];EBV BZLF1抑制IRF7在IFN-α/β啟動子上的轉錄活性,抑制NF-κB的表達[29];BZLF1還可誘導細胞信號轉導抑制因子3(Suppressor of cytokine signaling 3, SOCS3)的產生來抑制JAK/STAT信號傳導;EBV miR-BART16靶向干擾素途徑所需的轉錄輔助共激活因子環磷腺苷效應元件結合蛋白(cAMP response element binding protein,CREB)來抑制IFN-I信號傳導[30];EBV BILF4靶向IRF7下調IFN-α的表達水平[29];EBV miR-BART20-5p和miR-BART8可通過靶向STAT1影響干擾素-γ的信號傳導[30]。EBV的免疫逃逸策略也針對NLR3家族含吡咯結構域的炎性小體,EBV miRNAs可直接靶向NLRP3,降低其表達水平,或通過間接靶向IL-1受體,從而抑制促炎因子IL-1的信號傳導[30];EBV BART-6-3p靶向IL6R,抑制IL-6的信號轉導[30]。此外,還有部分病毒蛋白拮抗RLRs信號通路,如EBV miR-BART6-3p可直接與RIG-I mRNA結合,并在轉錄后抑制RIG-I mRNA的轉錄,從而抑制RLR介導IFN的產生[30]。
2.3 HCMV逃避宿主固有免疫應答反應的方式 HCMV作為一種普遍存在的條件致病病原體,在大多數健康個體中呈無癥狀感染,而在免疫缺陷個體,如獲得性免疫缺陷綜合征(Acquired immune deficiency syndrome, AIDS)患者、器官移植患者和老年人中會引起嚴重的并發癥。HCMV在感染宿主細胞時,HCMV病毒蛋白通過抑制cGAS-STING途徑中關鍵接頭蛋白的激活,進而抑制干擾素及炎性細胞因子的產生,最終促進自身的復制。例如,HCMV UL31通過與胞漿和胞核中的cGAS互作阻礙了DNA和cGAS的結合,從而使cGAMP的產生減少[31];HCMV UL83 N端結構域可與cGAS相互作用,導致cGAS-STING-IRF3信號通路受損引起IFN-β表達水平顯著降低,HCMV UL83和IFI16的相互作用抑制了IFI16的寡聚化,從而抑制細胞因子的轉錄激活,且有證據表明HCMV UL83下調了IFI16的DNA結合能力,并與HCMV的主要即刻早期啟動子(Major immediate early promoter, MIEP)形成復合物促進HCMV感染早期基因(Immediate early gene, IE)的表達,在感染早期啟動病毒轉錄[32];HCMV UL97可以阻斷IRF3和CBP互作,從而抑制IFN-β啟動子活性;HCMV UL48被報道以一種依賴于DUB的方式抑制STING、TRAF6、TRAF3、IL-1受體相關激酶1(Interleukin-1 receptor-associated kinase 1, IRAK1)、IRF7的表達,進而抑制其下游的信號傳導[33];研究發現,HCMV IE86(也稱為IE2,由UL122編碼)可通過蛋白酶體途徑降解STING,從而降低STING蛋白水平的表達,但對STING mRNA轉錄水平的表達無影響[34];HCMV IE86還抑制了NF-κB與IFN-β、IL-6、IL-8和RANTES啟動子區域的結合進而抑制上述細胞因子的基因轉錄[34]。在293T細胞中,HCMV UL82與STING互作,破壞了STING向高爾基體的易位,進而抑制TBK1和IRF3的激活[35]。HCMV US9可與MAVS和STING相互作用,通過阻止STING的二聚化和MAVS的激活,抑制細胞因子及炎性因子的表達[36]。HCMV UL26通過ISG化自身的誘餌蛋白來減少其他病毒ISG酰化蛋白的積累[37];HCMV UL26也可通過阻斷IKK復合體的磷酸化抑制NF-κB的活化,但ISG化的UL26則失去了這個功能[37]。此外,HCMV miRNA也在逃避宿主固有免疫應答過程中發揮著作用。HCMV miR-UL-112-3p被證明能夠抑制HCMV糖蛋白gB和gH與TLR2結合,抑制NF-κB介導的促炎細胞因子的產生[30]。HCMV miR-US5-1和miR-UL112-3p抑制IKKα和IKKβ的mRNA表達水平,從而抑制NF-κB的激活[30];HCMV miR-UL112-1也可通過抑制IL-32來下調炎癥因子的表達[30]。
2.4 KSHV逃避宿主固有免疫應答反應的方式 KSHV與淋巴細胞瘤、淋巴增殖性疾病以及其他腫瘤的發生發展密切相關[11]。最近大量研究表明,KSHV的多種蛋白和miRNA參與逃逸機體的固有免疫應答反應。如,KSHV ORF52結合并抑制cGAS的酶活性,阻礙cGAMP的產生[38]。KSHV潛伏期相關核抗原(Latency-associated nuclear antigen, LANA)與cGAS結合,阻止TBK1的下游蛋白的激活[32]。KSHV vIRF1直接與STING結合并阻止TBK1的募集[33]。KSHV ORF63與NLRP1結合干擾了炎性小體的形成和炎性細胞因子的產生[39]。KSHV去泛素化酶ORF64蛋白通過阻止NLRP1的泛素化和激活來抑制NLRs介導的信號通路的激活[40]。KSHV ORF64及其同源物MHV68 ORF64以去泛素化酶依賴的方式抑制STING依賴的信號轉導。KSHV ORF45通過與TBK1競爭結合IκB激酶ε(Inhibitor of NF-κB kinase ε, IKKε)來阻止IRF7的磷酸化和激活[41]。KSHV ORF8編碼的蛋白K-bZIP可抑制IRF3下游的信號轉導。KSHV ORF36作用機制類似于HSV-1 VP16。此外,KSHV miRNA也在逃避宿主抗病毒固有免疫應答機制中擔當著重要角色。如KSHV miR-K12-11靶向IKKε,抑制NF-κB和IRF3的激活。KSHV miR-K12-3和miR-K12-7靶向轉錄因子C/EBPβ,它抑制IL-6和IL-10的產生并促進病毒重新激活[30];KSHV miR-K12-5和miR-K12-9分別靶向MyD88和IRAK1下調它們的表達水平,從而抑制TLRs介導的細胞因子的激活[30];腫瘤壞死因子樣微弱凋亡誘導受體(TNF-related weak inducer of apoptosis receptor, TWEAKR)是促炎細胞因子的受體,KSHV miR-K10靶向TWEAKR下調促炎細胞因子的表達[30]。
2.5 其他人類皰疹病毒逃避宿主固有免疫應答反應的方式 與前面幾種人類皰疹病毒相比,VZV、HHV-6和HHV-7逃避宿主固有免疫反應的報道較少。帶狀皰疹是水痘初次感染后VZV重新激活的臨床表現。鑒于其可在宿主康復后長期潛伏的特性,那么VZV必然有其逃避宿主免疫反應的方式。目前,關于VZV和cGAS-STING通路之間的相互作用的研究甚少。現有文獻報道,HSV-1 ICP4的同源物VZV IE62可抑制TBK1介導的IRF3的磷酸化[3];VZV IE62通過阻斷IRF3的S396、S398和S402位的磷酸化抑制IRF3的二聚化[42];VZV ORF47以激酶依賴性的方式在IRF3 S396位點上阻止其磷酸化和同源二聚化[3];研究發現,在293T細胞中外源性的VZV ORF61以Ring-finger依賴性的方式特異性地靶向磷酸化的IRF3,并將其重新定位后被蛋白酶體所降解,但ORF61還可抑制IκBα的泛素化,進而抑制NF-κB信號的傳遞,該過程有助于IRFs信號的傳遞[3]。
玫瑰皰疹病毒(Roseoloviruses)包括HHV-6和HHV-7,其中HHV-6又分為HHV-6A和HHV-6B兩型。它們不僅會導致常見的兒童疾病如嬰兒玫瑰紅斑和嬰兒發熱性癲癇等,在免疫功能正常的人群中,它們與包括多發性硬化癥和阿爾茨海默氏癥在內的許多神經系統疾病有關。在293T細胞中,外源性HHA-6A/6B IE1可通過抑制IRF3的激活和核轉位,阻止其下游干擾素刺激基因及細胞因子的表達,逃避宿主的免疫應答[3]。
3 展 望
HHV與人類免疫系統經過數百萬年的共進化,形成了一種獨特的平衡狀態。探究清楚人類皰疹病毒逃避宿主免疫級聯反應的方式,有助于將來更好地研制出針對該病毒的免疫制劑,減輕其對人類健康和公共衛生帶來的危害[3]。本文總結了幾種人類皰疹病毒感染機體后所啟動的固有免疫級聯反應,并根據現有的研究成果討論了這些病毒通過干擾信號通路中的一些適配器分子的功能逃逸宿主免疫應答反應。人類皰疹病毒長期潛伏在宿主體內,因對大多數人所造成的疾病不嚴重或不發病而常常被忽視;但對特殊人群易感,一旦發病,危害比較嚴重。隨著對皰疹病毒結構及功能的深入研究,人們越來越清晰地認識到,人類皰疹病毒在宿主細胞免疫防御的各個階段均能夠通過影響免疫監視網絡的正常傳遞逃避機體的免疫級聯反應,并且與多種人類疾病密切相關。因此,闡明人類皰疹病毒與宿主細胞免疫應答反應之間的關系將為皰疹病毒的相關研究及其抗病毒藥物的研發提供寶貴的思路。
利益沖突:無
引用本文格式:牛宇輝,李向茸,馮若飛.人類皰疹病毒逃逸宿主抗病毒固有免疫應答機制的研究進展[J].中國人獸共患病學報,2022,38(1):62-68.DOI:10.3969/j.issn.1002-2694.2021.00.172
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
