凝固浴對再生膠原纖維結構與性能的影響
2022-09-22 14:29:08丁長坤岳程飛蘇杰梁閆旭煥程博聞
紡織學報 2022年9期
關鍵詞:力學性能
杜 璇, 丁長坤, 岳程飛, 蘇杰梁, 閆旭煥, 程博聞
1. 天津工業大學 材料科學與工程學院, 天津 300387; 2. 天津工業大學 天津市先進纖維與儲能技術重點實驗室, 天津 300387; 3. 天津科技大學, 天津 300457)
膠原(Col)是哺乳動物體內的重要結構蛋白,大多分布于皮膚、肌腱、骨、軟骨等部位中,具有獨特的三螺旋結構[1-3],具備生物相容性和生物可降解性優良以及免疫原性低等特點,在生物醫學工程等領域有著廣泛的應用[4]。其中,再生膠原纖維是可吸收手術縫合線的主要品種之一。
迄今,科研人員對膠原纖維的結構和性能進行了相關研究[5-6]。在傳統纖維成形技術中,濕法紡絲和干濕法紡絲應用最為普遍[7-8]。其中,凝固浴無疑是影響膠原纖維成形、結構和性能的重要因素之一。已報道的膠原纖維成形的凝固浴有丙酮[9-10]、硫酸鈉(Na2SO4)溶液[11]、碳酸鈉(Na2CO3)溶液[12]、磷酸鹽(PBS)緩沖液、氯化鈉(NaCl)溶液和聚乙二醇(PEG)溶液[13-14]等,其凝固成形時間分別為3~5 min(丙酮)、2 h(Na2SO4)、20 s(Na2CO3)、15 min(NaCl和PEG(相對分子質量為8 000)混合液)和48 h(PBS緩沖液),制備的纖維的斷裂強度差別亦較大(20~180 MPa(丙酮)、0.4~0.7 cN/dtex(Na2CO3)和154 MPa左右(PBS緩沖液))[15]。可以看出,目前所報道的凝固浴組成多種多樣,纖維凝固成形時間長短不一,而纖維的性能亦有所差別。
目前,對再生膠原纖維凝固成形系統的研究報道還比較少。為此,本文選擇4種(丙酮、NaCl溶液、PEG溶液、PBS緩沖液)使膠原紡絲液脫水及凝固速度不同的溶液作為凝固浴,運用干濕法紡絲技術制備再生膠原纖維,系統研究凝固條件對纖維微纖結構和力學性能的影響,總結不同凝固浴和成形條件下的纖維成形規律,為制備結構可控的醫用可吸收手術縫合線等膠原纖維材料提供參考。
1 實驗部分
1.1 實驗材料與儀器
材料:膠原(提取自牛肌腱),天津市賽寧生物工程技術有限公司;聚乙二醇(PEG,相對分子質量分別為4 000、6 000、8 000、10 000)、氯化鈉(NaCl)、磷酸鹽(PBS)緩沖液、氫氧化鈉、鹽酸,分析純,天津市風船化學試劑有限公司;冰醋酸、丙酮、氨水,分析純,天津市科密歐化學試劑有限公司。
儀器:干噴濕法紡絲裝置,實驗室自制;DW-I系列無極調速電動攪拌器,鞏義市予華儀器有限責任公司;TGL-16M型臺式高速冷凍離心機,湖南湘儀離心機儀器有限公司;LLY-06系列電子單纖維強力儀,萊州市電子儀器有限公司;S-4800型冷場發射掃描電子顯微鏡,日本日立公司;D8 DISCOVER型X射線衍射儀,德國布魯克公司;Nicolet iS50型傅里葉變換紅外光譜儀,美國賽默飛世爾科技有限公司。
1.2 再生膠原纖維的制備
再生膠原纖維紡絲原液的制備:以濃度為0.5 mol/L的冰醋酸溶液為溶劑,配制質量分數為1.5%的膠原紡絲原液,為避免溶解時膠原發生熱變性,溶解全程使溫度保持在4 ℃左右;膠原充分溶解后,于4 ℃下離心脫泡獲得膠原紡絲原液。
再生膠原纖維的制備:用自制干噴濕法紡絲裝置,將紡絲原液以0.5 mL/min的速度擠入凝固浴(氣隙高度為5~10 mm),溶液細流經過脫溶劑固化成形后獲得凝膠態膠原初生纖維;使用去離子水將初生纖維沖洗3~5次,再于室溫下以5 g砝碼懸吊拉伸,自然風干得到再生膠原纖維。凝固浴分別為丙酮[16](丙酮、氨水、去離子水按一定體積比例配制)、NaCl溶液(0.5、0.8、1.0、1.5 mol/L)、PEG溶液(質量分數為20%,溶于5 mg/mL的PBS緩沖液中)、PBS緩沖液(10 mg/mL,pH值為7.4),體積均為120 mL。本文實驗用1 mol/L NaOH和HCl調節NaCl溶液和PEG溶液的pH值進行相關分析。為方便敘述,4種凝固浴所得纖維分別記為丙酮-Col纖維、NaCl-Col纖維、PEG-Col纖維以及PBS-Col纖維。
1.3 結構表征與性能測試
使用電子單纖維強力儀測試再生膠原纖維的力學性能,夾距為10 mm,拉伸速率為10 mm/min,溫度為25 ℃,相對濕度為75%。每個試樣測試10次,取平均值。
1.3.2 形貌觀察
使用冷場發射掃描電子顯微鏡觀察樣品的表面、剖面與斷面形貌。測試前對樣品進行干燥與噴金處理,加速電壓為10 kV。
1.3.3 結晶性能測試
通過X射線衍射儀對干燥后的纖維進行結晶性能分析,具體測試條件:Cu靶為放射源,電流為30 mA,電壓為40 kV,掃描速度為5 (°)/min,掃描范圍為5°~40°。
1.3.4 化學結構表征
使用傅里葉變換紅外光譜儀對樣品的化學結構進行測試。取少量纖維與KBr粉末一起放入研缽中混勻磨細,壓成薄片制樣,測試范圍為4 000~400 cm-1,分辨率為2 cm-1。
2 結果與討論
2.1 纖維力學性能分析
圖1示出丙酮-Col纖維的力學性能曲線,依次以去離子水體積、氨水體積以及凝固時間為變量進行分析。丙酮會顯著降低水的介電常數[17],導致膠原分子脫水和聚集,凝固的速度相對較快,因此,可以加入適量水調節紡絲原液中的溶劑(去離子水)與凝固浴中的凝固劑(丙酮)之間的雙擴散速度。由圖1(a)可以看出:當去離子水的添加量較低時,雙擴散過程很快,導致纖維內部空隙較多,微纖堆積不夠緊密,纖維斷裂強度較低;隨著去離子水添加量的增加,雙擴散速度放緩,再生膠原纖維脫水變慢,纖維結構變得均勻,使得纖維的斷裂強度提高;當去離子水添加量為0.4 mL時,所得再生膠原纖維的斷裂強度最高為1.22 cN/dtex。但去離子水添加量持續增加,會導致雙擴散過于緩慢,以致于在相同時間內纖維成形程度低,使紡出的纖維力學性能變差。
目前,繼電保護裝置整機生產測試主要停留在手動測試或借助商用測試儀(如博電、昂立)和自主開發的測試儀進行的半自動測試階段。無論哪種測試方案,整個測試過程都過于依賴測試工程師的參與:測試工程師要不斷根據保護裝置的類型選擇測試線,并選擇相關功能進行測試。缺點是測試過程復雜,勞動強度較大,測試時間稍長。為此,本文深入研究智能電網保護裝置生產過程的各個子環節,通過對生產測試子過程的不斷集成及優化,生產過程大數據的綜合利用,整機測試儀的技術改進,氣動系統的靈活控制等,設計了繼電保護裝置整機智能測試系統,實現了繼電保護裝置的智能化大生產測試。
從圖1(b)可以看出,再生膠原纖維的力學性能隨凝固浴中氨水添加量的增加呈現出先上升后降低的趨勢,當氨水添加量為2~4 mL時,此時pH值更接近膠原的等電點[18],膠原分子脫水及聚集、凝固速度較快,所得再生膠原纖維斷裂強度較高。由圖1(c)可知,纖維的斷裂強度隨著凝固時間的延長先升高后降低,凝固時間太短使纖維成形慢,纖維內部膠原分子間作用力小,力學性能不高,而凝固時間太長則又使纖維斷裂強度下降。綜上所述,丙酮凝固浴的最佳條件為:去離子水添加量0.4 mL,氨水添加量2 mL,凝固時間3 min。
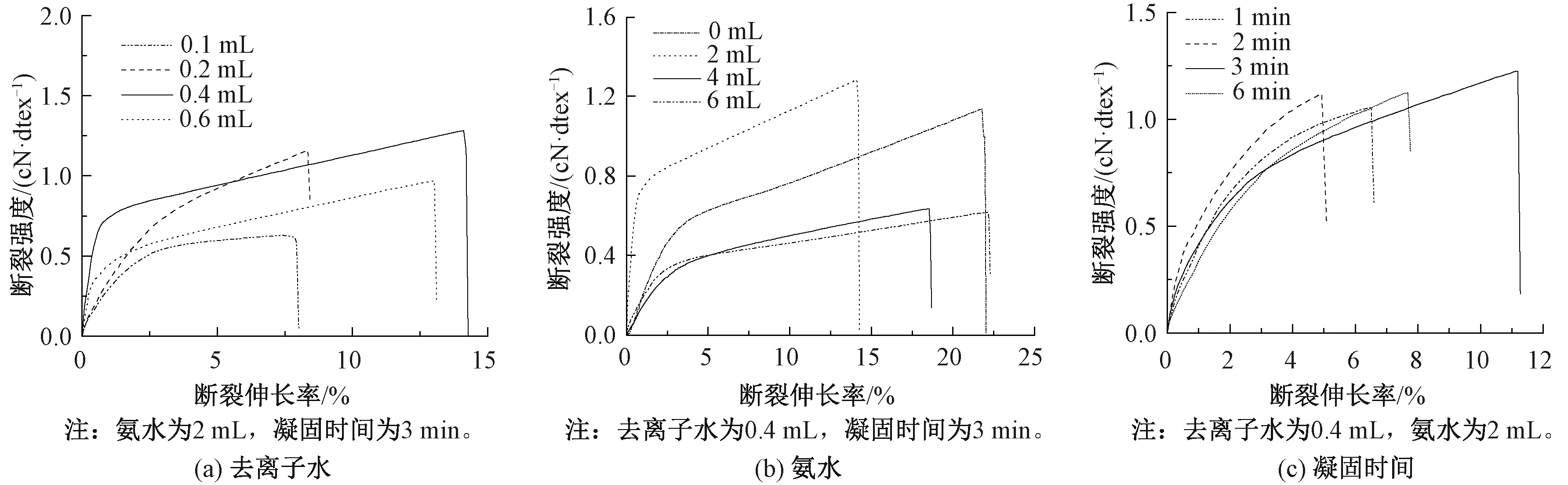
圖1 丙酮-Col纖維的力學性能
圖2示出NaCl-Col纖維的力學性能曲線,依次以NaCl濃度、pH值以及凝固時間為變量進行分析。由圖2(a)可看出,NaCl濃度的增加會使纖維的斷裂強度呈現出先升高后降低的趨勢。當NaCl濃度較低時,膠原溶液細流脫水較慢,造成纖維成形困難、強度差。隨著NaCl濃度的增加,鹽離子會使膠原分子表面的靜電作用力變大,分子間通過靜電作用更易聚集,使得膠原微纖自組裝過程加快,最終纖維力學強度得到提升。但當凝固浴中的NaCl超過一定濃度時,纖維的斷裂強度反而下降。這可能是因為纖維中一定量的NaCl殘留會使膠原分子及微纖的排列和堆積密度減小,微纖間的相互作用力下降[19],纖維力學性能變差。結合圖2(b)、(c)可以看出,當NaCl濃度為0.8 mol/L、pH值為7.4、凝固時間為10 min時,再生膠原纖維斷裂強度最高可達0.62 cN/dtex。
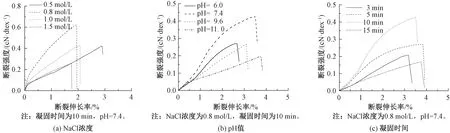
圖2 NaCl-Col纖維的力學性能
圖3示出PEG-Col纖維的力學性能曲線,依次以PEG相對分子質量、pH值以及凝固時間為變量進行分析。由圖3(a)可以看出,以PEG相對分子質量為8 000的溶液為凝固浴所得纖維的斷裂強度最優,達到0.43 cN/dtex。原因可能是短鏈的低相對分子質量的PEG 不僅比長鏈的高相對分子質量的PEG更易使膠原分子自組裝產生短原纖,還會隨機雜亂排列在膠原分子之間,影響膠原分子之間的相互作用和膠原微纖排列的規整程度,導致纖維強度較低。而PEG相對分子質量為8 000時正好可以在多肽螺旋結構有限的空間內促進膠原分子的運動和微纖的有序排列[13]。綜合圖3(b)、(c)可以看出,當PEG相對分子質量為8 000、pH值為7.4、凝固時間為8 min時,PEG-Col纖維力學性能較好。

圖3 PEG-Col纖維的力學性能
圖4示出不同凝固時間的PBS-Col纖維的力學性能曲線。可以看出,與前3種凝固浴相比,PBS緩沖液中再生膠原纖維的成形時間要長得多。這主要是因為組成PBS緩沖液的無機鹽的濃度很低,膠原溶液表面水化層的脫除速度很慢。當凝固時間為2 h時,纖維斷裂強度較低;當凝固時間為24 h時,纖維斷裂強度達最優為1.08 cN/dtex。較長的凝固時間使得膠原分子可以緩慢地進行自組裝,所形成微纖的有序、規整程度較好。但隨著凝固時間的進一步延長,纖維的斷裂強度卻有所下降,這可能是因為纖維內部殘留的更多的無機鹽引起的。
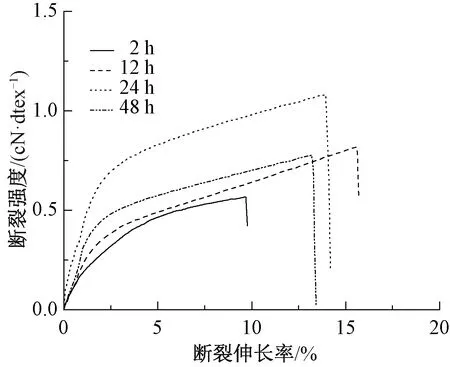
圖4 PBS-Col纖維的力學性能
2.2 纖維形貌分析
圖5示出2.1節得到的力學性能最優的丙酮-Col、NaCl-Col、PEG-Col和PBS-Col纖維的表面、剖面和斷面SEM形貌。

圖5 再生膠原纖維SEM照片
由圖5(a)可見,所有纖維表現出相似的表面形貌。丙酮-Col纖維和PEG-Col纖維表面較為光滑,且丙酮-Col纖維表現出更明顯的致密原纖化結構[20]。而NaCl-Col纖維和PBS-Col纖維因為有鹽的殘留使纖維中微纖堆積致密程度下降,表面稍顯粗糙。
由圖5(b)可以看出,丙酮-Col纖維內部的微纖較為細小,其沿著纖維軸向有序排列且堆積成緊密的聚集體,微纖間有很強的相互作用力,纖維的斷裂強度最高。而NaCl-Col纖維和PEG-Col纖維內部的微纖尺寸較大,排列較為疏松,微纖間隙較大;且NaCl-Col纖維可能因為NaCl的存在導致微纖尺寸有較大差異,PEG-Col纖維微纖較為均勻但微纖間連接仍然較少,這些原因導致這2種纖維的斷裂強度均較低。相比較,PBS-Col纖維的微纖尺寸也較大且均一性強,排列規整緊密,使得其斷裂強度要高于NaCl-Col纖維和PEG-Col纖維,但仍低于丙酮-Col纖維。圖5(c)的纖維斷面形貌呈現出相似的變化規律。
2.3 纖維結晶性能分析
圖6示出再生膠原纖維的XRD曲線。可見:在2θ為7°~8°處出現1個比較尖銳的小峰,反映的是膠原分子間的距離;第2個峰出現在20°左右,是1個較寬的饅頭峰,是纖維內部的結構層次漫散射造成的[21]。

圖6 再生膠原纖維XRD曲線
根據布拉格方程,NaCl-Col纖維(2θ=7.2°)、PEG-Col纖維(2θ=7.3°)和PBS-Col纖維(2θ=7.3°)的第1個衍射峰向左有微弱偏移,表明膠原分子間距變大,而丙酮-Col纖維(2θ=7.8°)的膠原分子間距較小,排列緊密而有序,這與纖維斷面的形貌觀察結果一致。
2.4 纖維化學結構分析
圖7示出再生膠原纖維的紅外光譜圖。可知,再生膠原纖維存在典型的酰胺Ⅰ帶、Ⅱ帶、Ⅲ帶和酰胺A帶、B帶的特征峰。在4 000~2 500 cm-1范圍內,3個吸收峰依次對應酰胺A帶、酰胺B帶以及—CH2的反對稱彎曲振動。1 635 cm-1處對應膠原的酰胺Ⅰ帶特征峰,該振動頻率通常受到肽鏈側基的影響,而酰胺Ⅱ帶(1 550 cm-1)對膠原螺旋結構不敏感。酰胺Ⅲ帶的吸收峰出現在1 241 cm-1處[22-23]。

圖7 再生膠原纖維紅外光譜圖
由圖7還可以看出,酰胺A帶和酰胺I帶2個特征峰并未發生明顯偏移,說明膠原的三股螺旋結構保持完好,即不同凝固浴所得纖維均完整地保留了膠原的特征結構,其可以發揮膠原的一系列生物學功能。對于PEG-Col纖維出現的差異性,認為是PEG分子在三股螺旋之間有限的空間內平行排列,導致紅外光譜圖中出現了PEG分子的特征峰[15,24]。
3 結 論
本文以丙酮、NaCl溶液、聚乙二醇(PEG)溶液、磷酸鹽(PBS)緩沖液為凝固浴,通過干濕法紡絲技術制備了再生膠原(Col)纖維,并對其進行結構表征及性能測試,得到如下主要結論。
1)丙酮會顯著降低水的介電常數,導致膠原分子脫水和聚集,凝固的速度相對較快。NaCl溶液和PBS緩沖液均通過脫除膠原表面的水化層使其析出,且二者都可使膠原表面的靜電作用力變大而易于聚集,但PBS緩沖液因鹽的濃度過低導致其脫水速度很慢。PEG及其相對分子質量大小會影響膠原分子之間的相互作用。
2)丙酮-Col纖維的微纖較為細小,排列有序程度高且堆積緊密,微纖間有很強的相互作用力,纖維斷裂強度最高。PBS-Col纖維的微纖尺寸較大且均一性強,排列較為緊密,斷裂強度次之。NaCl-Col和PEG-Col纖維微纖尺寸亦較大且有一定間隙,排列較為疏松,微纖間相互作用力低,二者斷裂強度較低。4種凝固浴所得纖維均完整地保留了膠原的三股螺旋結構。
猜你喜歡
材料與冶金學報(2022年2期)2022-08-10 09:15:46
云南化工(2021年11期)2022-01-12 06:06:14
山東冶金(2019年3期)2019-07-10 00:54:00
中國鑄造裝備與技術(2017年3期)2017-06-21 11:33:46
中國塑料(2016年6期)2016-06-27 06:34:16
西安工程大學學報(2016年2期)2016-06-05 12:25:17
中國塑料(2015年12期)2015-10-16 00:57:14
中國塑料(2015年9期)2015-10-14 01:12:26
中國塑料(2015年4期)2015-10-14 01:09:18
焊接(2015年9期)2015-07-18 11:03:53
