葡萄籽原花青素及其柔性納米脂質體的抗氧化及對酪氨酸酶活性的抑制作用*
2022-10-11 01:22:08趙聲蘭
中國藥業 2022年19期
關鍵詞:質量
竇 晨,丁 雄,潘 蕊,趙聲蘭,程 欣,3△
(1.云南中醫藥大學中藥學院,云南 昆明 650500;2.云南省高校外用給藥系統與制劑技術研究重點實驗室,云南 昆明 650500;3.云南省傣醫藥與彝醫藥重點實驗室,云南 昆明 650500)
人體黑色素的合成受黑色素生成途徑中的多種黑素體蛋白和酶的調控[1],包括酪氨酸酶、酪氨酸酶相關蛋白1和多巴色素互變異構酶等。酪氨酸酶可催化2個黑色素生成的限速步驟,即酪氨酸羥基化為3,4-二羥基苯丙氨酸(L-DOPA)和將L-DOPA氧化為多巴醌[2],故黑色素的產生主要依賴于酪氨酸酶的表達和激活[3]。生理劑量的自由基在黑色素生成的最后階段能加速5,6-二羥基吲哚和吲哚醌之間的反應,從而刺激黑色素的生成,增加皮膚的色素沉著[4]。葡萄籽原花青素(GSP)是從葡萄籽中提取的多酚化合物,是天然、低成本的抗氧化劑,具有抗氧化[5]、清除自由基[6]等藥理學活性,但分子結構中的不飽和鍵降低了其儲存的穩定性[7]。將GSP包載于柔性納米脂質體(FL)中,可增加其穩定性和功能性。本研究中探討了GSP及GSP-FL的抗氧化活性,以及對酪氨酸酶活性的影響,為制備具有美白功效的化妝品制劑提供參考。現報道如下。
1 儀器、試劑與細胞
1.1 儀器
OSB-2100型旋轉蒸發儀(倍捷科技<上海>有限公司);90Plus型納米激光粒度儀(美國布魯克海文儀器公司);Sorvall ST 8R型高速冷凍離心機,Multiskan型酶標儀(美國Thermo Fisher Scientific公司);Discovery DV215CD型分析天平(美國奧豪斯公司,精度為0.0001 g);Scientz-IID型超聲波細胞破碎儀(寧波新芝生物科技股份有限公司);PI80H型超聲波處理器(德國Elma公司)。
1.2 試劑
GSP(天津市尖峰天然產物研究開發有限公司,批號為002200604028,純度不低于95%);大豆卵磷脂(批號為1112J021),四甲基偶氮唑鹽(MTT,批號為715F051),均購自北京索萊寶科技有限公司;膽固醇(批號為624J033),脫氧膽酸鈉(批號為909P021,純度大于99%),均購自北京蘭杰柯科技有限公司公司;L-酪氨酸(批號為RH221350),酪氨酸酶(批號為R002875),三吡啶三吖嗪(TPTZ,批號為R012773),均購自上海凜恩科技發展有限公司;L-DOPA(美國Sigma公司,批號為WXBD0797V);曲酸(上海阿拉丁生化科技股份有限公司,批號為12002059);1,1-二苯基-2-三硝基苯肼(DPPH)自由基(上海源葉生物有限公司,批號為S28J12M138964);2,2'-聯氮-雙-3-乙基苯并噻唑啉-6-磺酸(ABTS,梯希愛<上海>化成工業發展有限公司,批號為KZTCI-AE);曲拉通X-100(TritonX-100,美國Amresco公司,批號為21B1756838);2-苯基-4,4,5,5-四甲基咪唑啉-1-氧基-3-氧化物(PTIO,九鼎化學<上海>科技有限公司,批號為YCLQJNY);七 水 合 硫 酸 亞 鐵(FeSO4,批 號 為20200918),過氧化氫(H2O2,批號為20210201),均購自廣東光華科技股份有限公司;水為超純水。
1.3 細胞
小鼠永生化黑色素細胞系Melan-a細胞來源于廣州醫科大學,由云南中醫藥大學藥劑教研室培養。
2 方法與結果
2.1 GSP-FL的制備(薄膜分散-超聲法)
稱取卵磷脂250 mg、膽固醇17 mg,精密稱定,分別溶于15 mL氯仿;稱取脫氧膽酸鈉15 mg,溶于5 mL甲醇,超聲溶解。將上述3種溶液置圓底燒瓶中,混勻。40℃水浴條件下以旋轉蒸發儀去除有機溶劑,直至燒瓶內壁形成干燥、透明的均勻薄膜。稱取GSP 80 mg,溶于6 mL水,置圓底燒瓶中,40℃水浴條件下繼續旋轉30 min,冰浴條件下超聲(功率為150~180 W,頻率為20~25 kHz)處理5 min,0.22 μm微孔濾膜濾過,4℃條件下密封保存。在前期優化制備工藝的基礎上制備GSP-FL[8],采用超速離心法測定,其包封率為(70.79±0.02)%,載藥量為(1.16±0.01)%,粒徑為(156.47±7.03)nm,多 分 散系數為0.26±0.01,Zeta電位為-(42.35±4.00)mV。
2.2 GSP及GSP-FL的抗氧化活性
2.2.1 DPPH自由基清除率測定
取DPPH自由基2 mg,精密稱定,置50 mL容量瓶中,用無水乙醇定容,即得質量濃度為0.04 mg/mL的DPPH自由基溶液,避光保存,現配現用。采用80%乙醇溶解維生素C(VC),采用無水乙醇溶解GSP及GSP-FL,配制成質量濃度分別為3.13,6.25,12.5,25,50 μg/mL的待測試樣溶劑。于96孔板[9]中加入各反應液,50 μL待測試樣溶劑和150 μL DPPH自由基溶液為A0,150 μL DPPH自由基溶液及50 μL待測試樣為A1,50 μL待測試樣和150 μL待測試樣溶劑為A2。37℃條件下反應30 min,采用酶標儀于519 nm波長處測定吸光度。VC為陽性對照,A2為空白對照,以空白柔性納米脂質體(EL)排除脂質體對吸光度的干擾。DPPH自由基清除率(%)=[1-(A1-A2)/A0]×100%。采用SPSS 26.0統計學軟件進行回歸分析,并計算半數抑制濃度(IC50)。
結果見圖1。可見,在3.13~50 μg/mL質量濃度范圍內,VC,GSP,GSP-FL對DPPH自由基的清除率均逐漸增加;對DPPH自由基的IC50分別為(15.277±2.392)μg/mL、(9.791±0.615)μg/mL、(9.271±0.424)μg/mL。GSP與GSP-FL表現出比VC更好的DPPH自由基清除活性。EL對DPPH自由基的活性無影響。

圖1 GSP,GSP-FL,VC對DPPH自由基的清除率Note:Compared with Vc,aP<0.05(for Fig.1-4).Fig.1 Scavenging rates of GSP,GSP-FL and VC on the DPPH free radical
2.2.2 ABTS+自由基清除率測定[10]
取過硫酸鉀13.40 mg和ABTS 38.40 mg,精密稱定,分別置25 mL容量瓶中,用純水充分溶解并定容,置燒杯中1∶1混勻,室溫避光放置12 h,即得ABTS+自由基貯備液;使用前用純水稀釋10~20倍,直至吸光度為0.70±0.02,即得ABTS+自由基工作液。采用80%乙醇溶解VC,采用無水乙醇溶解GSP及GSP-FL,配制成質量濃度分別為1.56,3.13,6.25,12.5,25 μg/mL的溶液。于96孔板中加入各反應液,75 μL待測試樣溶劑和75 μL ABTS+自由基工作液為A0,75 μL ABTS+自由基工作液及75 μL待測試樣為A1,75 μL待測試樣和75 μL待測試樣溶劑為A2。室溫下反應6 min,采用酶標儀于734 nm波長處測定吸光度。VC為陽性對照,A2為空白對照。ABTS+自由基清除率(%)=[1-(A1-A2)/A0]×100%。
結果見圖2。可見,在1.56~25 μg/mL質量濃度范圍內,VC,GSP,GSP-FL對ABTS+自由基的清除率均逐漸增加;對ABTS+自由基的IC50分別為(12.387±1.79)μg/mL、(10.722±1.80)μg/mL、(10.403±2.33)μg/mL。GSP,GSP-FL,VC的ABTS+自由基清除活性相似。EL對ABTS+自由基的活性無影響。
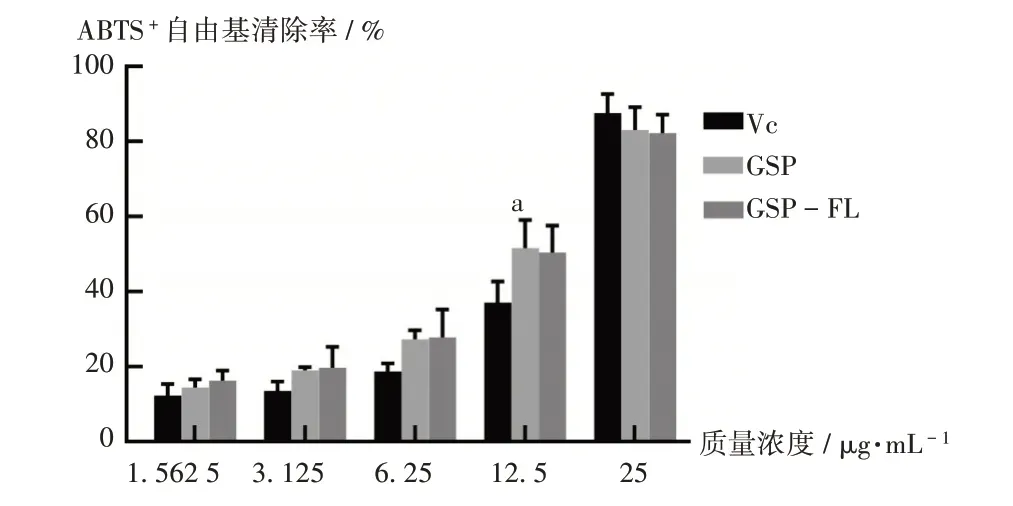
圖2 GSP,GSP-FL,VC對ABTS+自由基的清除率Fig.2 Scavenging rates of GSP,GSP-FL and VC on the ABTS+free radical
2.2.3 PTIO自由基清除率測定[11]
取PTIO自由基10 mg,精密稱定,置50 mL容量瓶中,用純水定容,即得PTIO自由基工作液。用純水溶解VC,GSP,GSP-FL,配制成質量濃度分別為0.0125,0.025,0.05,0.1,0.2,0.4 mg/mL的溶液。于96孔板中加入各反應液,40 μL純水和160 μL PTIO自由基工作液為A0,160 μL PTIO自由基工作液及40 μL待測試樣為A1,40 μL待測試樣和160 μL純水為A2。37℃條件下靜置30 min,采用酶標儀于557 nm波長處測定吸光度。以VC為陽性對照。PTIO自由基清除率(%)=[1-(A1-A2)/A0]×100%。
結果見圖3。可見,在0.0125~0.4 mg/mL質量濃度范圍內,VC,GSP,GSP-FL對PTIO自由基的清除率均逐漸增加;對PTIO自由基的IC50分別為(0.097±0.001)mg/mL、(0.220±0.056)mg/mL、(0.187±0.037)mg/mL。GSP與GSP-FL表現出對PTIO自由基的清除活性,但效果不如VC。GSP-FL質量濃度為0.0125~0.1 mg/mL時,對PTIO自由基的清除率顯著高于VC(P<0.05)。EL對PTIO自由基的活性無影響。
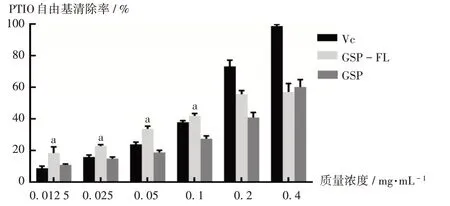
圖3 GSP,GSP-FL,VC對PTIO自由基的清除率Fig.3 Scavenging rates of GSP,GSP-FL and VC on the PTIO free radical
2.2.4 鐵離子(Fe3+)還原能力測定[10]
取TPTZ粉末7.8 mg,精密稱定,溶于2.5 mL 40 mmol/L鹽酸溶液中,超聲(功率為80 W,頻率為40 kHz)使其完全溶解,即得10 mmol/L TPTZ溶液;取27.029 mg氯化鐵,精密稱定,置10 mL容量瓶中,用純水溶解并定容,即得10 mmol/L氯化鐵溶液;將300 mmol/L醋酸鹽緩沖液、10 mmol/L TPTZ溶液和10 mmol/L氯化鐵溶液按10∶1∶1混勻,即得Fe3+抗氧化能力工作液。取FeSO450.59 mg,精密稱定,置25 mL容量瓶中,用純水溶解并定容,梯度稀釋成濃度分別為0.5,1,1.5,2,2.5,3,3.5 mmol/L的FeSO4標準溶液。在96孔板中加入不同濃度的FeSO4溶液20 μL,再加入180 μL Fe3+抗氧化能力工作液,在37℃條件下恒溫孵育6 min,采用酶標儀于593 nm波長處測定吸光度。以FeSO4溶液的濃度為橫坐標(X)、吸光度為縱坐標(Y)進行線性回歸,得標準曲線Y=1.009X+0.2084(R2=0.9927,n=7)。在96孔板中加入20 μL質量濃度分別為0.05,0.1,0.2 mg/mL的GSP,GSP-FL,VC溶液,再加入180 μL Fe3+抗氧化能力工作液,37℃條件下恒溫孵育6 min,采用酶標儀于593 nm波長處測定吸光度。樣品溶液對Fe3+的還原能力以FeSO4含量表示,以VC為陽性對照。根據標準曲線測定GSP,GSP-FL,VC溶液對Fe3+的還原能力。
結果見圖4。可見,在0.05~0.2 mg/mL質量濃度范圍內,GSP,GSP-FL,VC對Fe3+的還原能力均逐漸增強,但GSP和GSP-FL的還原能力不如VC。EL對Fe3+的還原能力無影響。
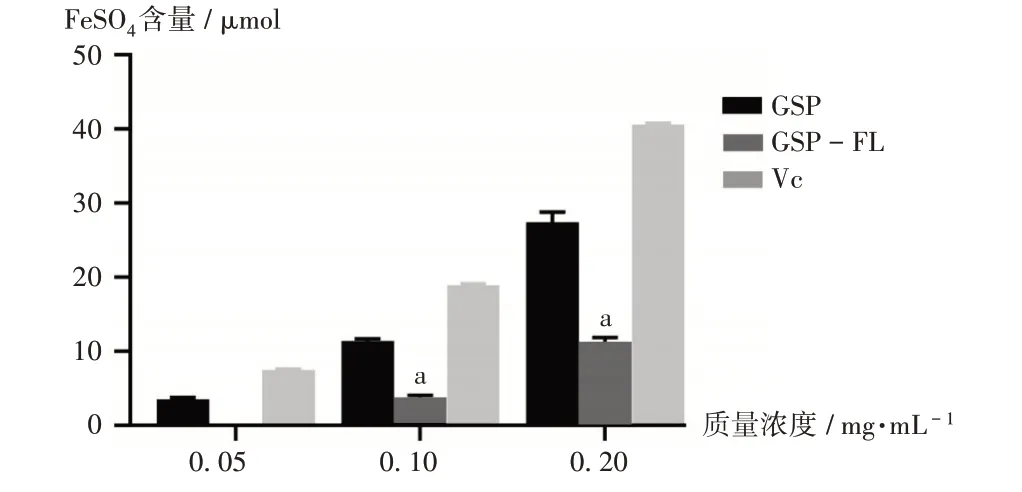
圖4 GSP,GSP-FL,VC對Fe3+的還原能力Fig.4 Reduction abilities of GSP,GSP-FL and VC on the Fe3+
2.3 GSP對H2O2誘導Melan-a細胞氧化損傷的保護作用
2.3.1 GSP及GSP-FL對Melan-a細胞的毒性(MTT法)
取處于對數生長期的Melan-a細胞,胰蛋白酶消化,離心(轉速為800 r/min)3 min,采用血細胞計數法調整細胞懸液的密度為7.5×105/mL,接種于96孔板,每孔100 μL,置細胞培養箱(溫度為37℃,CO2濃度為5%,下同)中培養24 h,棄去細胞培養基;每孔分別加入100 μL質量濃度分別為0.0125,0.025,0.05,0.1,0.2,0.4,0.8 mg/mL的GSP及GSP-FL溶液,每個質量濃度設5個復孔,重復試驗3次。另設陰性對照組(等量的細胞+細胞培養液)與空白對照組(等量的細胞培養液),于細胞培養箱中分別培養24 h和48 h。加入20 μL質量濃度為5 mg/mL的現配MTT溶液,于細胞培養箱中孵育4 h,棄去上清液。每孔加入150 μL二甲基亞砜,振搖10 min,充分溶解,采用酶標儀于490 nm波長處測定吸光度。細胞抑制率(%)=[1-(AGSP/GSP-FLA空白對照組)/(A陰性對照組-A空白對照組)]×100%。
結果見圖5。可見,與GSP-FL相比,給藥24 h后,GSP質量濃度為0.0125~0.8 mg/mL時的細胞毒性較強,且48 h時的細胞毒性更強。給藥24 h和48 h后,GSP-FL的平均IC50為(0.443±0.09)mg/mL和(0.137±0.00)mg/mL,顯著高于GSP的(0.140±0.40)mg/mL和(0.049±0.01)mg/mL(P<0.05)。GSP-FL降低了游離藥物的細胞毒性,提高了細胞的存活率。故推測將GSP包載于FL中可顯著增加藥物的安全性。EL質量濃度為0.8 mg/mL時,對Melan-a細胞的抑制率為(25.2±9.99)%,隨著其不斷稀釋,抑制率降低,故EL毒性較低。
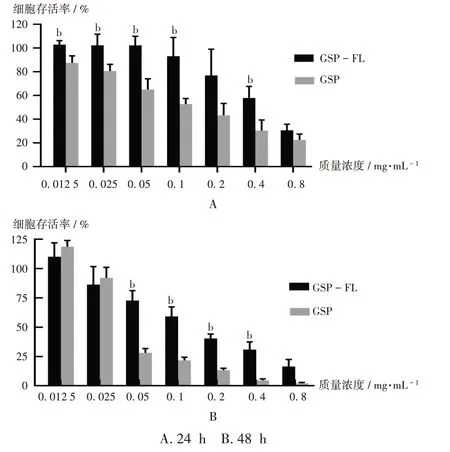
圖5 加入GSP和GSP-FL后Melan-a細胞的存活率(n=3)A.24 h B.48 hNote:Compared with those with GSP,bP<0.05.Fig.5 Survival rates of Melan-a cells after adding GSP and GSP-FL(n=3)
2.3.2 H2O2誘導Melan-a細胞氧化損傷模型的建立
取處于對數生長期的Melan-a細胞,用含有10%胎牛血清的1640完全培養液稀釋,直至細胞懸液的密度為5×104/mL,接種于96孔板,每孔100 μL,置細胞培養箱中培養48 h,棄去上清液,每孔加入100 μL 25,50,100,200,400,800 μmol/L的H2O2溶液,造模8 h,每個濃度設置5個復孔,重復試驗3次。另設陰性對照組(等量的細胞+細胞培養液)與空白對照組(等量的細胞培養液)。計算細胞抑制率。結果H2O2溶液濃度為25,50,100,200,400,800 μmol/L時,Melan-a細胞抑制 率 分 別 為(9.11±5.81)%、(16.69±9.60)%、(40.95±3.52)%、(63.18±4.64)%、(91.09±3.04)%、(93.21±2.26)%(n=3)。
2.3.3 GSP對H2O2誘導Melan-a細胞氧化損傷的保護作用(MTT法)[12]
前期試驗考察了150,250 μmol/L H2O2的作用,結果其濃度為250 μmol/L時,GSP組的保護作用與模型組相比,具有顯著差異(P<0.05)。故將細胞分為4組,即陰性對照組(等量的細胞+細胞培養液)、空白對照組(等量的細胞培養液)、模型組(250 μmol/L H2O2)、不同質量濃度(6.25,12.5,25,50 mg/mL)的GSP組。按2.3.2項下方法將Melan-a細胞均勻接種于96孔板,空白對照組僅加入等量培養液,培養24 h,棄去上清液。GSP組分別加入含不同質量濃度GSP的培養液100 μL,在空白對照組、陰性對照組和模型組中加入等量的培養液,培養24 h,棄去上清液。在模型組和GSP組中分別加入100 μL 250 μmol/L的H2O2溶液,在空白對照組、陰性對照組中加入等量培養液。將96孔板轉移至細胞培養箱中培養8 h,計算細胞抑制率。
結果見圖6。可見,與模型組比較,GSP質量濃度為12.5~50 mg/mL時,能顯著增加H2O2造成的氧化損傷細胞的存活率(P<0.05);但GSP質量濃度為50 mg/mL時,其保護作用低于25 mg/mL,推測在質量濃度為50 mg/mL時,H2O2對細胞仍有一定毒性。由于FL減少了H2O2與細胞的直接接觸,降低了藥物對細胞的刺激性。
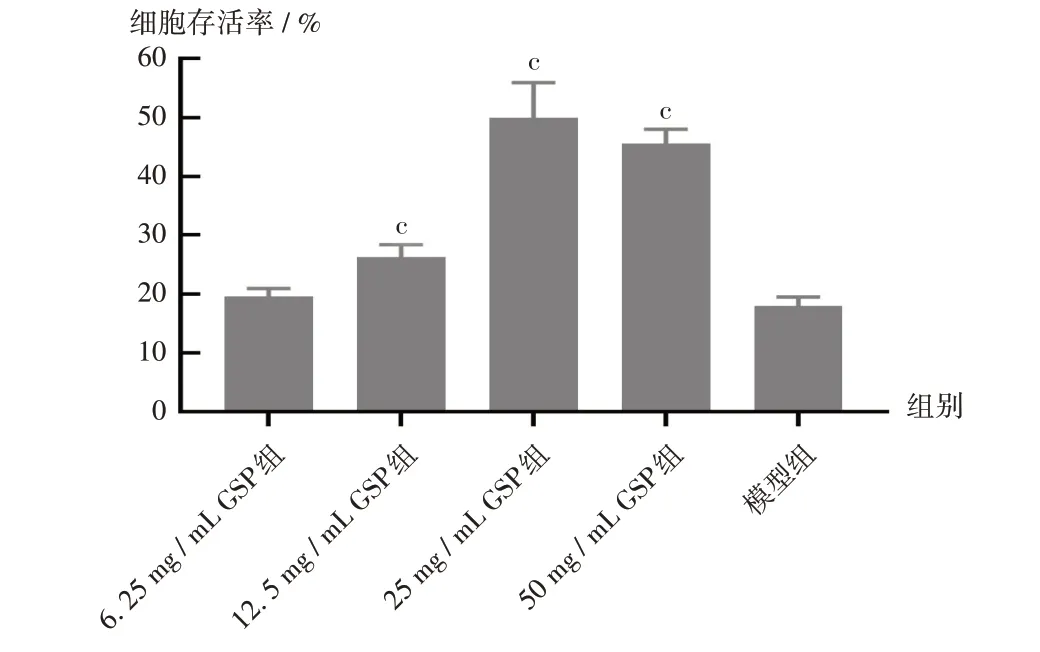
圖6 不同GSP質量濃度對氧化損傷Melan-a細胞存活率的影響Note:Compared with those in the model group,cP<0.05.Fig.6 Effects of GSP with different mass concentrations on the survival rates of oxidatively damaged Melan-a cells
2.4 GSP和GSP-FL對體外酪氨酸酶活性的影響
2.4.1 對酪氨酸酶單酚酶及二酚酶活性的影響
以2.0 mmol/L的酪氨酸溶液或2.0 mmol/L的L-DOPA為底物,按表1進行反應,采用96孔板,在200 μL測活體系中加入濃度為2.0 mmol/L的酪氨酸溶液(或L-DOPA)和磷酸鹽緩沖液,置37℃培養箱中預熱10 min,加入質量濃度為0.1,0.2,0.4,0.8 mg/mL的GSP,GSP-FL溶液各50 μL及質量濃度為0.003,0.006,0.012,0.025,0.05 mg/mL的曲酸溶液50 μL,加入質量濃度為150 U/mL酪氨酸酶溶液50 μL,置37℃培養箱中繼續反應15 min,采用酶標儀于475 nm波長處測定吸光度。抑制率(%)=[(A1-A2)-(A3-A4)]/(A1-A2)×100%。

表1 體外酪氨酸酶活性試驗反應液組成(μL)Tab.1 Composition of reaction solution for tyrosinase activity test(μL)
結果顯示見圖7。可見,曲酸質量濃度低于0.05 mg/mL時,對酪氨酸單酚酶及二酚酶活性均有抑制作用;GSP和GSP-FL對酪氨酸酶活性的抑制作用均隨質量濃度的升高而降低。GSP-FL對酪氨酸單酚酶的IC50約為GSP的2倍,對酪氨酸二酚酶的IC50約為GSP的3.5倍。詳見表2。
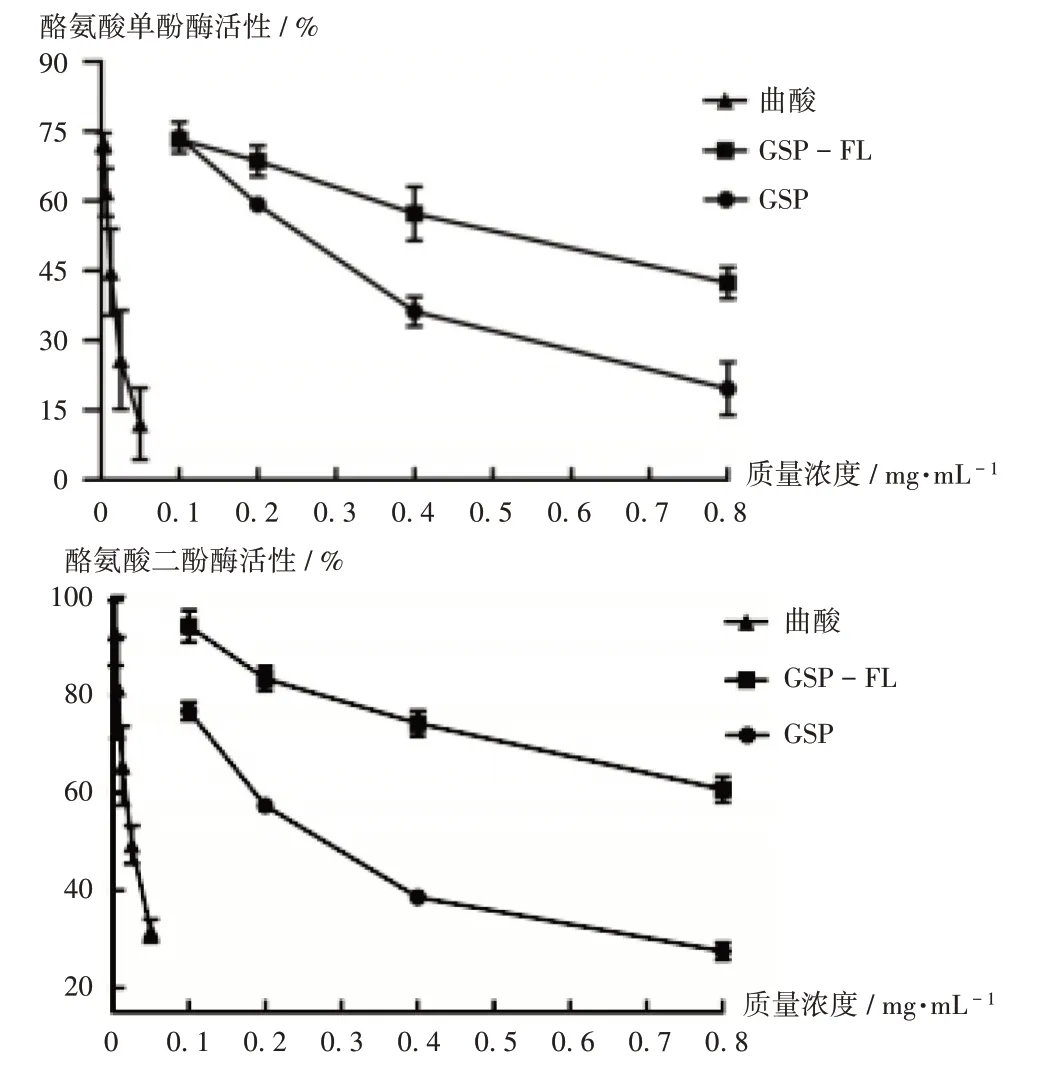
圖7 GSP和GSP-FL、曲酸對酪氨酸酶單酚酶及二酚酶的活性作用Fig.7 Effects of GSP,GSP-FL and kojic acid on the monophenolase and diphenolase activities of tyrosinase
表2 對酪氨酸酶單酚酶及二酚酶作用的IC50(±s,mg/mL)Tab.2 IC50 of GSP,GSP-FL and kojic acid on the monophenolase and diphenolase of tyrosinase(±s,mg/mL)

表2 對酪氨酸酶單酚酶及二酚酶作用的IC50(±s,mg/mL)Tab.2 IC50 of GSP,GSP-FL and kojic acid on the monophenolase and diphenolase of tyrosinase(±s,mg/mL)
注:與曲酸比較,dP<0.05。表3和表4同。Note:Compared with those with kojic acid,dP<0.05(for Tab.2-4).
試劑GSP GSP-FL曲酸酪氨酸二酚酶0.294±0.011.057±0.03d 0.024±0.01酪氨酸單酚酶0.243±0.000.503±0.08d 0.010±0.00
2.4.2 酶促動力學反應
固定底物L-DOPA溶液的濃度為2 mmol/L,分別配制濃度為20,40,60,80,100 U/mL的酪氨酸酶溶液及質量濃度為0.05,0.1,0.2,0.4,0.8,1.6 mg/mL的GSP溶液,按表1進行反應,測定GSP對酪氨酸酶的抑制機制,得到酶活性隨酶量變化的關系圖(圖8),酶活性對酶量是一條相對的直線,隨著待測樣品溶劑質量濃度的升高,酶活性減少。提示GSP是通過抑制酶活性而不是減少有效酶量來降低對L-DOPA的催化效率的。
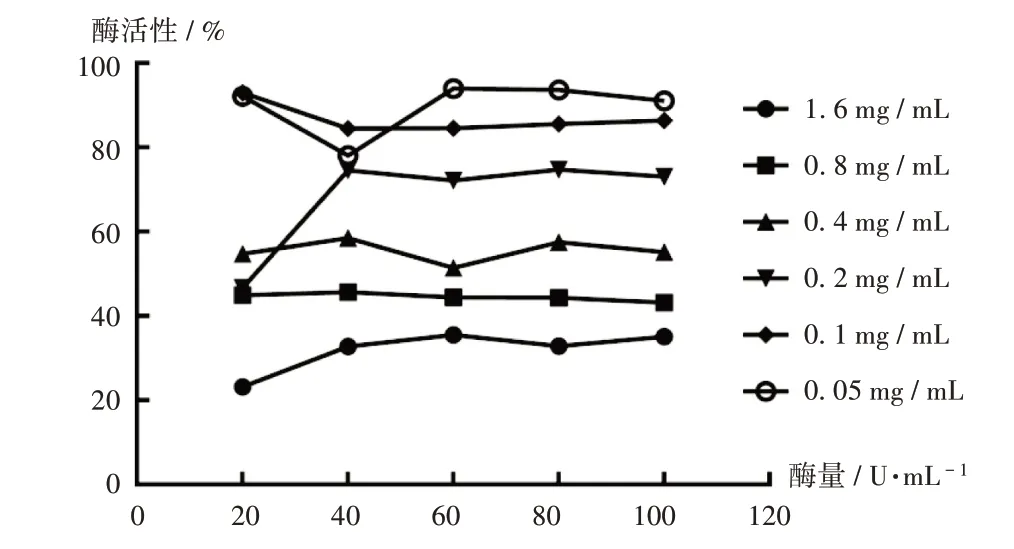
圖8 GSP對酪氨酸酶的抑制效應機制Fig.8 Mechanism of inhibitory effect of GSP on tyrosinase
2.5 GSP和GSP-FL對細胞內酪氨酸酶活性的影響
2.5.1 Melan-a細胞胞內酪氨酸酶抑制率[12-14]
取對數生長期的Melan-a細胞,胰蛋白酶消化,調整細胞懸液的密度為2.5×105/mL,并接種于6孔板中,培養24 h。加入含不同質量濃度GSP、GSP-FL及曲酸的培養基,另設陰性對照(等量的細胞+細胞培養液)。培養24 h和48 h,棄去培養基,加入500 μL 1% TritonX-100溶液,靜置5 min。用細胞刮刀刮下每孔中的細胞,置冰面充分裂解1 h,冷凍,離心(轉速為14000 r/min)10 min。取上清液100 μL,加入96孔板中,置37℃培養箱中預熱10 min。加入3 mmol/LL-DOPA溶液100 μL,繼續反應1 h,采用酶標儀于475 nm波長處測定吸光度,每個濃度設置3個復孔,同時設置空白孔(100 μL 1%TritonX-100和100 μL 3 mmol/LL-DOPA),重復3次 試 驗。酪 氨 酸 酶 活 性(%)=(AGSP/GSP-FL/曲酸-A空白對照組)/(A陰性對照組-A空白對照組)×100%。
結果見表3。可見,隨著作用時間的增加,GSP及GSP-FL對酪氨酸酶活性的抑制作用均逐漸增強。24 h后,在質量濃度為0.8 mg/mL的GSP及GSP-FL作用下,Melan-a細胞胞內酪氨酸酶活性顯著低于曲酸(P<0.05);48 h后,在質量濃度為0.4 mg/mL的GSP及GSP-FL作用下,Melan-a細胞胞內酪氨酸酶活性顯著低于曲酸(P<0.05)。

表3 GSP及GSP-FL、曲酸對細胞內酪氨酸酶活性的影響(n=3)Tab.3 Effects of GSP,GSP-FL and kojic acid on the activity of intracellular tyrosinase(n=3)
2.5.2 Melan-a細胞胞內黑色素含量
取對數生長期的Melan-a細胞,胰蛋白酶消化,調整細胞懸液的密度為2.5×105/mL,并接種于6孔板中,培養24 h。加入含不同質量濃度GSP-FL及曲酸的培養基,另設陰性對照組(等量的細胞+細胞培養液),培養24 h或48 h,棄去培養基。加入500 μL 1%TritonX-100溶液,靜置5 min,用細胞刮刀刮下每孔中的細胞,置冰面上充分裂解1 h,冷凍,離心(轉速為14000 r/min)10 min。取離心后的細胞黑色素沉淀,加入100 μL 1 mol/L NaOH溶液(含10%二甲基亞砜)中,80℃條件下加熱1 h至完全溶解,采用酶標儀于405 nm波長處測定吸光度,重復3次試驗。黑色素含量(%)=(AGSP-FL/曲酸)/A陰性對照組×100%。
結果見表4。可見,隨著GSP-FL質量濃度的升高和作用時間的延長,黑色素的合成均減少。24 h后,GSP-FL質量濃度為0.8 mg/mL時,Melan-a細胞胞內黑色素含量顯著低于曲酸組(P<0.05);48 h后,GSP-FL質量濃度為0.4 mg/mL時,細胞胞內黑色素含量顯著低于曲酸組(P<0.05)。因藥物本身存在的顏色會干擾黑色素吸光度的測定,且無法通過試驗設計進行排除,故未對GSP進行測定。
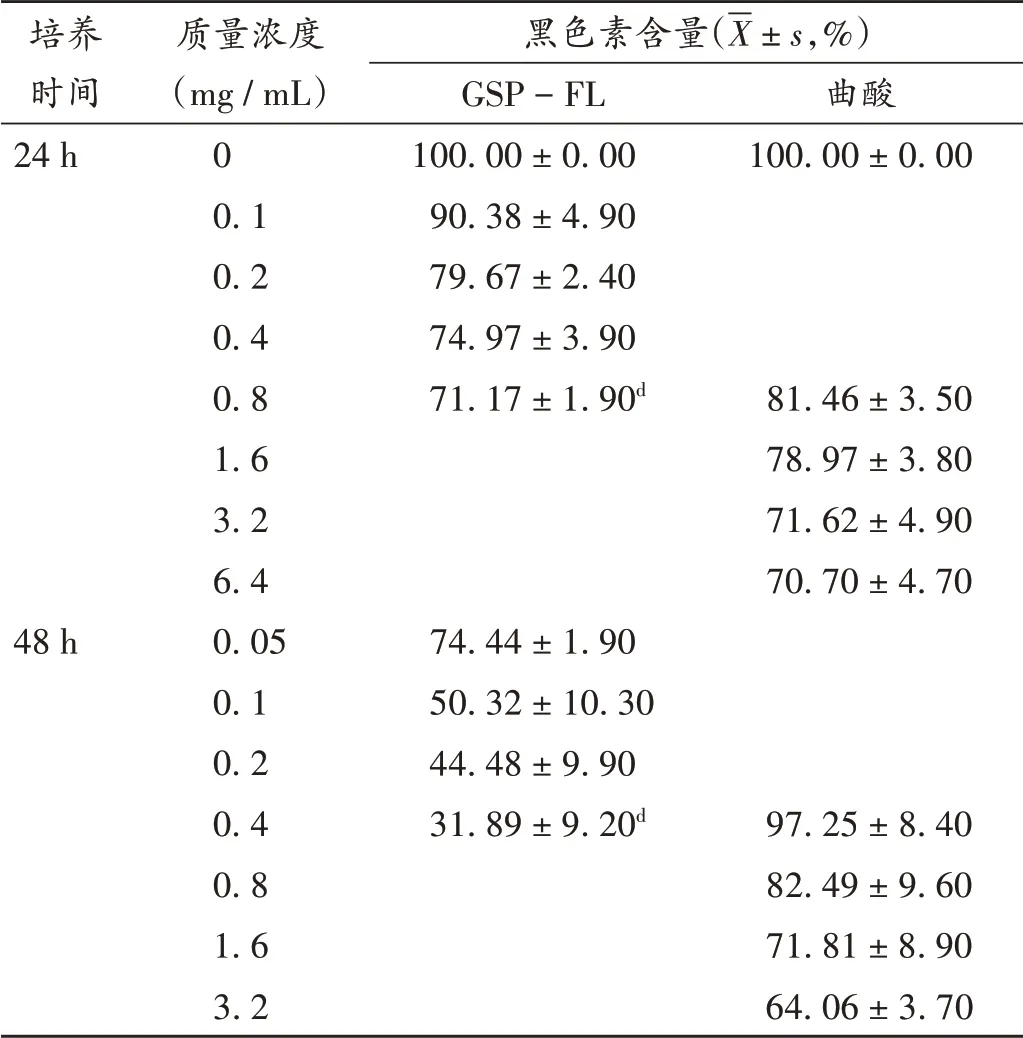
表4 GSP-FL對細胞內黑色素合成的影響(n=3)Tab.4 Effect of GSP-FL on the synthesis of intracellular melanin(n=3)
3 討論
多數體外抗氧化的檢測方法僅能評價化合物清除某一種自由基的能力,且各有優、缺點,各種方法聯用才能更好地反映某化合物的抗氧化活性。本研究中通過測定GSP及GSP-FL對DPPH,ABTS+,PTIO自由基的清除能力及對Fe3+的還原能力,綜合評價其抗氧化活性,結果表明GSP及GSP-FL均具有良好的抗氧化活性,對DPPH和ABTS+自由基的IC50均低于VC。早有研究報道,GSP是比VC和維生素E琥珀酸乙酯更強的自由基清除劑[15]。GSP能顯著增加H2O2氧化損傷細胞的存活率,降低細胞的氧化損傷程度,但其作用機制需作進一步研究。
體外酶學試驗結果顯示,GSP和GSP-FL均表現出對酪氨酸單酚酶及二酚酶的抑制作用;但GSP-FL對酪氨酸單酚酶及二酚酶的抑制率顯著低于GSP,推測可能是由于FL外殼對藥物的保護作用,在缺乏破乳劑的情況下,藥物未突釋出來。MTT試驗結果顯示,GSP會抑制正常Melan-a細胞的增殖,將GSP包載于FL中,顯著降低了藥物的細胞毒性。推測可能是由于脂質體的磷脂雙分子層外殼較安全,可減少藥物與細胞的直接接觸,從而降低了藥物對細胞的刺激性。在細胞試驗中,GSP及GSP-FL表現出對Melan-a細胞內酪氨酸酶活性、黑色素生成的抑制作用。皮膚美白劑的功能應在安全的藥物劑量下減少細胞內黑色素的合成,不能因為過多的黑色素細胞死亡而使局部的皮膚顏色變淺,影響美觀,甚至在無黑色素的保護作用下,導致皮膚曬傷[16-17]。GSP-FL在抑制細胞胞內酪氨酸酶活性和黑色素生成的同時,降低了細胞毒性,減少了黑色素細胞的死亡。
綜上所述,GSP-FL具有清除自由基、抗氧化、抑制酪氨酸酶活性和黑色素生成的作用,相比游離GSP和GSP-FL還能降低對正常細胞的毒性。有望將GSP-FL進一步開發成凝膠劑、乳膏劑等具有美白功效的化妝品制劑,但美白是多重酶、多種因素共同作用的結果,其藥理作用和藥效機制還需進一步驗證。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
