材料基因工程加速新材料設計與研發
2022-10-13 09:59:06孫志梅王冠杰張烜廣周健
北京航空航天大學學報 2022年9期
關鍵詞:數據庫
孫志梅 王冠杰 張烜廣 周健
(1. 北京航空航天大學 材料科學與工程學院, 北京 100083;2. 北京航空航天大學 集成電路科學與工程學院, 北京 100083)
先進材料是國家工業的支柱,在傳統“試錯-糾錯”研發模式下,材料學家基于自身知識儲備和認知能力,通過反復迭代的試錯-糾錯改進材料性能,實現新材料的設計與研發。 隨著新一輪工業革命和互聯網時代的到來,新材料的研發速度嚴重滯后于材料性能需求速度,按需逆向設計和精準控制性能已成為新材料設計的必然趨勢。 20世紀末,美國興起組合材料學(combinatorial materials science, CMS)[1],通過并行合成和高通量表征技術,實現了新材料的快速制備和篩選;21 世紀初,美國和歐洲部分國家提出的集成計算材料工程(integrated computational materials engineering, ICME)[2]將不同時間尺度和空間尺度的多種材料模擬方法相結合,在新材料設計領域取得了突破。 如今,隨著計算機和人工智能技術的飛速發展,材料基因工程(materials genome engineering,MGE)被視為實現材料科學技術飛躍和新材料高效研發與設計的基礎,是新材料研發的加速器。
材料基因工程是受人類基因組計劃(human genome project, HGP)的啟發而建立的。 在生物學中,基因是一組編碼信息,被視為生物體生長和發育的藍圖,而在材料領域,基因可被看作決定其宏觀性能的微觀特征單元。 作為基于數據驅動的科學發展第四范式,材料基因工程將高通量計算和設計、高通量制備、高通量表征、材料數據庫和人工智能相結合,大大縮短了材料研發周期、降低了研發成本,從而快速研發出滿足日益增長的性能需求的新材料。
2002 年,美國賓夕法尼亞州立大學Liu[3-4]首次提出材料基因的概念,并于2005 年設立材料基因基金會。 2011 年,美國正式發布提升美國全球競爭力的材料基因組計劃(materials genome initiative, MGI)[5],確立了面向未來的集成計算、實驗和數據庫的材料研發新模式。 MGI 構建了包含上百萬條先進能源材料的計算模擬結果材料數據庫,并提出了計算機輔助材料研發、模塊化的材料模擬體系、開放式的材料高性能數據庫以及多尺度計算融合等研究方向。 歐盟也相繼提出了“新材料發現NOMAD”計劃、德國推出了工業4.0戰略、俄羅斯推出“2030 年前材料與技術發展戰略”、中國提出了“材料基因工程”等一系列政策將新材料探索和材料創新設計與研發作為首要發展目標。
本文首先介紹了國內外材料基因工程領域常用的高通量計算模擬軟件和框架。 其次,從材料數據來源、多類型數據庫和數據標準方面介紹了目前常用的材料數據庫。 然后,總結了機器學習方法在材料學中的熱點應用,重點介紹了筆者團隊自主開發的多尺度集成可視化的高通量自動計算和數據管理智能平臺ALKEMIE 研究進展。 最后,總結提出了材料基因工程未來的重點發展方向。
1 材料高通量計算方法
科學發展經歷了如圖1 所示的4 個過程:從文藝復興時期實驗主導的經驗范式、以經典力學和熱力學為主導的理論模型范式、基于分子動力學和密度泛函理論的計算科學范式,到如今的大數據驅動的科學研究范式[6]。 近年來,美國、歐洲、日本和中國等國家科研人員在數據驅動研發模式的推動下,開發了一系列用于材料計算基礎設施的高通量計算框架和“即插即用”功能完善的高通量計算軟件,如表1 所示。 2011 年,在美國國家科學技術委員會、能源部和教育部支持下,加利福尼亞大學伯克利分校勞倫斯伯克利國家實驗室Jain 等[7]主導開展了材料基因組項目(materials project, MP),該項目開發了4 款分別用于材料建模、材料計算模擬、自動糾錯和服務器部署的高通量計算分析軟件。 其中,FireWorks 用于構建材料高通量計算模擬框架,解決高通量材料計算過程中多任務間的依賴關系和任務間的參數及數據傳遞;Custodian 用于高通量計算過程中的自動糾錯;Pymatgen 通過抽象的Python 對象解析材料結構對稱性,自動分析不同尺度計算模擬軟件的輸入參數和結果,工作原理如圖2 所示;Atomate則實現了完整的高通量流程及數據存儲和服務器的配置,如圖3 中能帶計算工作流所示。 該項目以其獨特的命令行操作方式為高通量計算奠定了軟件基礎。

圖2 Pymatgen 高通量軟件工作原理[7]Fig.2 Principle of Pymatgen high-throughput software[7]

圖3 Atomate 中高通量能帶計算流程[7]Fig.3 High-throughput computational workflow of band structure in Atomate[7]

表1 材料高通計算軟件和框架發展現狀Table 1 Software and frameworks of material high-throughput calculation

圖1 科學發展的四個范式[6]Fig.1 Four paradigms of science[6]
2012 年,同屬美國材料基因組項目的杜克大學Curtarolo 等[8]基于Python2 開發了適用于第一性原理計算的高通量計算軟件AFLOW-π。 該軟件針對高通量第一性原理計算,集成了數據實時反饋、錯誤控制、數據管理和歸檔等功能,可用于實現能帶結構、態密度、聲子色散、彈性特性、復介電常數、擴散系數等高通量計算,并針對性地優化了緊束縛的哈密頓量(tight-binding Hamiltonians,TBH)計算和數據分析流程。
丹麥科技大學Larsen 團隊[9]開發了材料批量化計算平臺原子模擬環境(atom simulation environment, ASE), 該軟件由于缺少工作流程、計算參數和結果的自動糾錯功能,并非完整意義上的高通量計算。 而隨著版本的迭代,科研人員為其進一步添加了可視化的用戶界面、多個材料軟件計算器、多種算法的分子動力學計算軟件和多種晶體結構優化算法及邊界條件,可以滿足不同用戶不同功能的計算需求。 目前,該平臺包含了ABINIT、CASTEP、CP2K、VASP 和LAMMPS 等常用的材料計算模擬軟件。
2016 年,瑞士洛桑聯邦理工大學Pizzi 等[10]開發了高通量計算引擎(automated interactive infrastructure and database for computational science,AiiDA),該軟件系統基于自動化、數據庫和開源共享理念,開發了支持數萬個材料計算任務并發運行的高通量算法。 材料科學家不僅關注計算模擬的輸入和輸出,更關注計算模擬過程中的精度及構型的變化是否準確,因此,該軟件保存了材料高通量計算中的子任務依賴關系,并自動跟蹤記錄所有計算和工作流程的輸入、輸出和中間元數據,以便在其開放式數據庫Materials Cloud 中查詢數據。
基于MP 發展的材料高通量計算基礎框架,比利時天主教魯汶大學Gonze 等[11]開發了基于多體微擾論的第一性原理高通量計算軟件Abipy;美國佛羅里達大學Mathew 等[12]發展了針對二維材料表面和異質結的高通量計算流程MPInterfaces(見圖4);英國倫敦國王學院Lambert等[13]發展了用于原子晶界的高通量計算框架Imeall 等。

圖4 二維材料表面和異質結的高通量計算軟件MPInterfaces 工作流程[12]Fig.4 Workflow for 2D material surfaces and heterojunctions in MPInterfaces software[12]
相比于國外材料基因工程的研究成果,中國高通量計算起步較晚,但在2016 年《中華人民共和國國民經濟和社會發展第十三個五年規劃綱要》中材料基因國家重點專項支持下,中國也涌現了多個成熟的材料高通量計算框架和軟件。
多尺度集成可視化的高通量自動計算和數據管理智能平臺(artificial learning and knowledge enhanced materials informatics engineering, ALKEMIE)是由筆者團隊Wang 等[14-15]基于Python 開源框架自主開發的中國第一個高通量自動流程可視化計算和數據管理智能平臺。 該平臺從設計出發吸取國外材料基因相關軟件的先進理念,克服了計算過程中可能遇到的兼容性差、接口不統一和功能拓展困難等問題,開發了包含材料高通量自動計算模擬、材料數據庫及數據管理、基于人工智能和機器學習的材料數據挖掘3 個核心理念的智能平臺。 ALKEMIE 平臺適用于數據驅動的材料研發,詳細內容見第4 節。
MIP(materials information platform)是由上海大學Yang 等[16]開發的適用于熱電材料高通量篩選的高通量計算軟件;MatCloud 是由北京邁高材云科技有限公司Yang 等[17]開發的第一性原理計算引擎,目前支持VASP 和ABINIT 等第一性原理高通量計算軟件;JAMIP 是由吉林大學Zhao等[18]開發的開源高通量集成軟件,該軟件利用人工智能算法在高通量計算的海量材料數據中智能尋求新材料和新原理。 中國材料基因工程高通量計算平臺(CNMGE)則是由國家超級計算天津中心開發的網頁版高通量集成計算平臺,該平臺可以集成多種不同的高通量計算軟件,包括ALKEMIE 高通量智算平臺、含能材料分子專用高通量篩選系統EM-Studio 和用于無機骨架材料的晶體結構解析與預測軟件(framework generator, FraGen)。
上述高通量計算軟件和框架,一方面為科研人員提供了快捷方便的自動計算工作流,可以在高性能超算中實現高效并行計算,顯著提升計算效率;另一方面避免了傳統試錯-糾錯法中可能出現的人為誤差,使得研究人員有更多的精力關注材料科學問題本身,而非高通量所涉及的技術難題。 近年來,高通量計算模擬已經在材料構型預測、材料結構穩定性和相穩定性預測、能源材料能量轉化效率、最優摻雜元素分級篩選等方面獲得了廣泛應用。 舉例來說,Curtarolo 等[19-20]通過高通量方法從435 個含d 電子的二元金屬間化合物中篩選出了283 個能量穩定化合物,其中273 個(96.5%)化合物最終獲得了實驗驗證;Hu 等[21]基于第一原理計算的層級篩選,從29 個過渡金屬元素中篩選出可以提高相變材料Sb2Te3性能的最佳摻雜元素Y、Sc 和Hg,除了有毒的Hg 元素,Y 和Sc 均獲得了實驗驗證;Gan 等[22]基于第一原理計算的高通量分級篩選,通過結構能量、聲子譜、力學穩定性和轉換效率等4 個篩選標準從21 060個候選光電材料中篩選出了78 個穩定化合物,且其中22 個化合物的性能優于當前太陽能電池材料GaAs。
2 材料數據庫研究進展
高通量計算大幅提高了計算模擬的效率,并產生了海量的數據,這些數據中既包含了有用的材料性質數據,又包含了大量重復的無效數據。而材料學中由于所研究的材料體系、成分、結構等的不同,材料測試、制備工藝和流程也不盡相同。對于不同用途材料,所關注的材料性能和關鍵指標也有差異,因此,在數據庫的構建過程中面臨數據存儲類型、數據庫的兼容性和泛化能力等一系列問題。 本文根據材料數據來源,將數據大致分為材料計算數據、材料測試數據和已發表的文本數據三大類。 本節將介紹目前國內外常用的材料數據庫和數據共享標準。
2.1 材料多類型數據庫
目前常用的材料數據庫如表2 所示。 ICSD是由萊布尼茨信息基礎設施研究所Belsky 等[23]構建的無機材料結構數據庫,收錄了自1913 年以來在1 600 個學術期刊發表的超過8 萬篇論文中的共計21 萬條晶體結構數據,覆蓋金屬、有機物、同素異形體等各種形態的材料體系;COD (crystallography open database) 是由英國劍橋大學Quirós 等[24]開發的包含超過700 萬個有機、無機、金屬有機化合物和礦物的晶體結構數據庫;MP Database 是由美國加利福尼亞大學伯克利分校勞倫斯伯克利國家實驗室Jain 等[7]建立的材料計算模擬專用數據平臺(見圖5),不僅收錄了材料結構數據,也收錄了元素性質、電子結構、彈性張量和能源轉換電極性能等數據;AFLOWLIB(automatic flow lib)是由美國杜克大學Curtarolo等[19]基于AFLOW-π 高通量軟件開發的材料計算數據庫,數據庫中收錄了6 400 余條熱力學相圖數據和超過45 萬個四元混合物的材料性質數據;Materials Cloud 是由瑞士洛桑聯邦理工大學Pizzi 等[10]開發的第一性原理計算元數據的數據庫,包括超過752 萬條第一性原理結構弛豫流程和納米多孔材料吸收和擴散相關的材料性質數據;OQDM(open quantum database for materials)是由美國西北大學Saal 等[25]開發的第一性原理計算熱力學數據庫,包含了數萬個二元、三元和四元相圖;NOMAD(novel materials discovery)是由歐洲馬克斯·普朗克學會Draxl 等[26]開發的歐洲最大的新材料共享數據庫,該數據庫包含了49 TB的各類材料數據;MatNavi 是由日本國家材料科學研究所Ogata 等[27]開發的多種材料數據的集合,包含聚合物數據庫(化學結構、加工、物理性質、NMR 光譜數據)、無機材料數據庫(晶體結構、相圖、物理性質)、金屬材料數據庫(密度、彈性模量、蠕變性質、疲勞特性)、電子結構計算數據庫等。
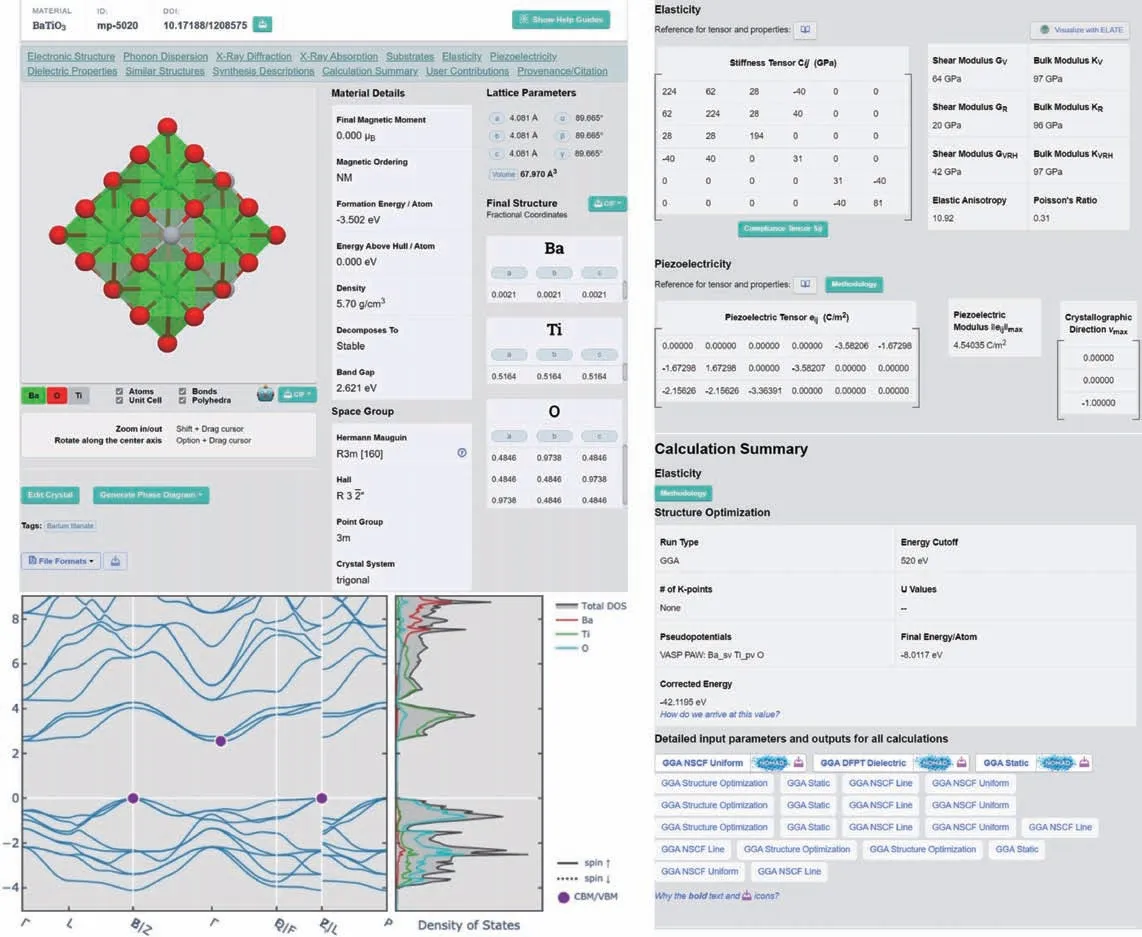
圖5 Materials Project Database 數據庫概況[7]Fig.5 Snapshot of Materials Project Database[7]

表2 材料多類型數據庫的發展Table 2 Development of multi-type databases of materials
除了上述通用的多類型材料數據庫,還有眾多針對材料某個特定領域的特色數據庫。 例如,美國佛羅里達大學開發的材料表面界面數據庫Materials Web; 美國國家標準技術研究所Choudhary 等[28]開發的贗勢數據庫JARVIS-DFT;丹麥科技大學Larsen 等[9]開發的二維材料、硒化物和硫化物數據庫(computational materials repository, CMR);北歐理論物理研究所Borysov 等[29]開發的三維有機晶體材料電子結構和帶隙數據庫(organic materials database,OMDB);美國斯坦福大學Hummelsh?j 等[30]開發的催化材料活化能數據庫CatApp;此外,還有ASM 出版社開發的熱處理數據表及應力應變和蠕變曲線材料數據庫(ASM alloy center database,ASMDB)、礦物材料數據庫(American mineralogist crystal structure database,AMSD)和儲氫材料數據庫(hydrogen storage materials database,HSMD)等。
中國目前也發展了多個大型材料數據庫共享平臺。 ALKEMIE-DB 是由筆者團隊Wang 等[14-15]基于高通量智能計算平臺ALKEMIE 開發的多類型材料數據庫(見圖6),該數據庫分為隱私數據庫和共享數據庫兩大類,根據數據類型進一步細分為含60 余萬組數據的晶體結構數據庫、含1 萬余條聲子能帶的聲子譜數據庫、含20 余萬組數據的深度學習贗勢數據庫、高通量計算工作流數據庫等。 MSDSN 是由國家統籌建設、北京科技大學實施完成的材料科學數據共享平臺,主要分為包含有色金屬材料和特種合金和微觀組織模擬的實驗數據庫、熱力學和動力學相關的計算模擬數據庫等。 Atomly 是由中國科學院物理研究所松山湖材料實驗室和懷柔材料基因平臺共建的材料計算數據庫,目前包含20 萬條材料結構數據和5 萬條相圖數據等。 MCDC(national materials corrosion and protection data center)是由國家科技部門建設的腐蝕防護數據平臺,包含環境數據、腐蝕數據、腐蝕檢測和腐蝕預測等。

圖6 ALKEMIE-DB 數據庫中高通量能帶和態密度計算結果可視化[14]Fig.6 Visualization of high-throughput band structures and density of states calculations in ALKEMIE-DB databases[14]
2.2 材料數據標準
隨著高通量計算、高通量實驗和超級計算機計算能力的發展,材料數據形成的數據海(data ocean)面臨著5 個重要的挑戰(“5V”特性):
1) 速率(velocity)。 新數據產生速率和舊數據更新速率越來越快,對數據的格式化存儲和快速讀寫提出了更高的要求。
2) 數據量(volume)。 材料數據以TB 量級不斷增加,需要可靠的數據存儲、高效的數據檢索和開放共享的可重復利用。
3) 多樣性(variety)。 材料數據的存儲形式、材料體系、測試和計算方法,以及數據蘊含的物理意義更加多樣化。
4) 真實性(veracity)。 數據的不確定性和可靠性決定了數據是否真實有效,進一步決定了數據挖掘和機器學習模型的精度和泛化能力。
5) 低價值密度(value)。 數據價值密度高低與數據總量大小成反比,數據量越大,數據價值密度越低。
如何在海量數據中分析預測數據隱藏的真實意義和價值是材料基因工程方法努力探索的主要方向。
為了解決上述問題,歐洲馬克斯·普朗克學會Draxl 等[26]提出了材料數據庫建設的FAIR Data 準則,即可發現(findable)、可獲取(accessible)、可互操作(interoperable)和可再利用(reusable),來提升材料數據的開源共享性。 歐洲用于材料設計的開放式數據庫集成團隊Andersen 等[31]提出了材料數據共享標準OPTIMADE(見圖7),該標準通過JSON 格式定義了材料數據共享的統一標準,目前大多數材料數據庫MP Database、AFLOWLIB、NOMAD、Materials Cloud 和ALKEMIEDB 等均提供了OPTIMADE 通用接口支持。
圖7 材料數據共享標準OPTIMADE 概況[31]Fig.7 Overview of materials data sharing standard of OPTIMADE[31]
中國中關村材料試驗技術聯盟于2019 年也提出了材料基因工程數據通則,將材料科學數據分為樣品信息、源數據(未經處理的數據)與衍生數據(經分析處理得到的數據)三大類,并從宏觀上定義了材料數據的通用性與專用性;北京科技大學材料基因工程北京市重點實驗室在2020 年MSDSN 數據庫中定義了材料科研數據DOI 編碼規則:“10. 11961/classification. project. date. sequence”,編碼中包含了材料數據分類號、項目支撐信息、注冊日期和流水號;北京航空航天大學ALKEMIE-DB 數據庫也發展了用于材料數據共享的唯一標識符:“alkemie. date. classification/user_defined_label. number.”,其中alkemie 為數據庫社區唯一標識,date 代表數據創建日期(精確到μs),classification 表示數據類別,user_defined_label 為用戶自定義字段,number 為數據唯一索引序號。
3 機器學習在材料設計中的應用
機器學習傳統上分為監督學習和非監督學習兩大類。 監督學習是指給算法一個數據集,對于數據集中的每一個樣本,都給出對應的映射(即標簽),算法的目的是給出更多的映射,得到更多的答案;而非監督學習常常被用于在大量無標簽數據中探索并發現規律。 進一步,監督學習分為處理連續值的回歸問題和處理離散值的分類問題。 目前,常用的機器學習算法已經有很多相關綜述,本文不再贅述。 隨著計算機科學和人工智能的發展,機器學習在材料結構設計、材料性能預測和材料分析圖像識別等領域扮演著越來越重要的角色。 本節主要介紹機器學習在材料性能預測、材料數據文本挖掘和機器學習原子間作用勢。
3.1 材料性能預測
機器學習在材料學中的應用主要包含格式化材料數據、機器學習模型訓練和高效材料性能預測3 個步驟,并在二維、光伏、催化、合金和熱電等材料中均獲得了顯著的研究成果。 近年來,瑞士洛桑聯邦理工大學Lin 等[32]通過高通量篩選和機器學習算法,從ICSD 和COD 數據庫的108 423個三維晶體構型中,通過對稱性和幾何算法篩選了1 036 個容易合成和789 個具有潛在可能性的層狀二維材料混合物;東南大學Lu 等[33]通過高通量篩選和機器學習描述符,從5 158 個候選材料中篩選了能量轉化效率高且帶隙在0.9 ~1.6 eV之間有機無機結合的鈣鈦礦光伏材料(HOIPS),并構建了可以高效預測帶隙值的結構描述符和機器學習模型,進一步將模型拓展到雙層HOIPS 中,成功從11 370 個混合物中預測了204 個無毒且穩定的光伏材料[34];北京科技大學Zhang 等[35]在銅合金體系中通過貝葉斯優化迭代方法,分別構建了誤差小于7%的硬度模型和誤差小于9%的電導率模型,并通過迭代優化設計了兼具優異力學和電學性能的Cu-Ni-Co-Si-Mg 合金。
3.2 材料數據文本挖掘
2018 年,瑞士聯邦理工學院Villars 等[36]通過計算機視覺和自然語言方法從已經正式發表的論文中自動識別有效材料結構和數據,并通過數據挖掘探索數據背后隱藏的物理模型和機理,構建了MPDS(materials platform for data science),近年來,該團隊首次從35 000 篇論文中解析了15 500個化學成分,并基于其中的2 330 個二元體系構建了機器學習分類模型;進一步從超過8 000 篇發表的論文中構建了2 800 個二元混合物的原子配位環境多面體分析算法(atomic environment types,AETs);隨后,從超過50 000 篇已發表論文中分析了290 000 個原子配位環境數據,將該算法模型拓展到了多元無機混合物中。 2019 年,Tshitoyan 等[37]發展了Word2vec 非監督機器學習模型,成功從330 萬材料文本中篩選了超過7 000 個候選的熱電材料。 除了材料計算模擬數據,在實驗合成方式上,Kononova 等[38]通過文本挖掘和自然語言處理方式從53 538 篇科學文獻數據中篩選了19 488 個無機金屬合成方式,包括材料成分、制備條件、化學平衡方程和反應過程,該數據庫為實驗中無機材料的制備過程提供了有力的數據支持。
3.3 機器學習原子間作用勢
材料計算模擬根據模擬時長和體系大小分為原子尺度、分子尺度、介觀尺度和宏觀尺度模擬,尺度越小模擬精度越高,尺度越大越接近真實體系,但是不同的模擬尺度采用的物理模型和近似原理不同,數據耦合非常困難,而數據驅動的機器學習方法被視為材料多尺度模擬的耦合劑。 經典大規模分子動力學常被用來模擬近似真實材料體系的服役性能,但可靠、精確的原子間勢函數的匱乏限制了其廣泛應用。 基于密度泛函原理(DFT)的第一性原理模擬具有精確的贗勢庫,但求解本征值所需的巨大計算量限制了該方法在大的原子尺度和時間尺度上的模擬,常用的VASP 僅限數百原子的體系。 因此,簡單方便地獲得適用于經典分子動力學的可靠勢函數非常重要。 隨著計算機技術、計算機視覺和材料基因理念的快速發展,通過機器學習結合大數據、高通量計算的方法擬合可靠的適用于經典分子動力學模擬的勢函數成為了研究熱點。
機器學習勢函數的發展主要經歷了原子個數受限的低維度勢函數和泛化能力強的高維度神經網絡勢函數2 個發展過程。 1995 年, Blank 等[39]開發了第一個基于統計學的勢函數模型,用于研究氫原子的分子動力學模擬;2009 年,Malshe等[40]進一步提出了通過神經網絡預測經典多體勢方程參數的模型。 但是,上述模型均不能改變輸入的原子個數,因此限制了機器學習勢函數的應用。
2011 年,Behler[41]提出了原子中心對稱函數,通過數學方程解析原子局域環境,構建了輸入原子個數不受限的高維度神經網絡模型。2018 年,Gastegger 等[42]發展了權重相關的對稱函數(wACSF),通過卷積神經網絡提升了模型的精度和實用性,但是由于局域近似,無法包含超過截斷半徑的原子長程相互作用。 2018 年,Yao等[43]提出了包含長程靜電作用和散射作用的HDNNP 方法,但是該方法并未獲得廣泛應用,一方面由于物理學中超過6 ~10 ? 的靜電作用通常對體系的整體影響較小,另一方面添加長程作用會急劇增加模型訓練成本,與其對精度的微小提升相比有待進一步優化。 筆者團隊Wang 等[14]開發了適用于相變材料Sb 單質的跨尺度機器學習勢函數PotentialMind,該勢函數模型與DFT 比較,對能量預測的精度達到98%,平均到每個原子上的能量誤差值小于0.045 eV/atom,對力的預測精度達到89%,該算法具有很強的擴展性和通用性,易于擴展到多元材料體系中。
機器學習勢函數方法一方面實現了具有第一原理精度且更大原子數體系和更長時間尺度的大規模分子動力學模擬,另一方面通過替代求解復雜多體薛定諤方程本征值,使得模擬速度提升2 ~3個數量級,目前,該方法已經在多個材料體系中獲得了應用。 例如,Sosso 等[44]發展了適用于二元相變存儲材料GeTe 的人工神經網絡勢函數,并實現了具有第一原理精度的4 096 個原子體系的大規模分子動力學模擬,通過模擬相變材料的多個淬火過程(100 ~300 ps),探究了淬火速度和模擬體系大小對GeTe 非晶結構的影響;Artrith和Urban[45]基于Fortran 語言開發了適用于鈣鈦礦TiO2的神經網絡勢函數軟件(atomic energy network, AENET),并面向科研人員開源使用,加速了機器學習勢函數方法在能源材料中的應用;Mocanu 等[46]通過高斯近似方法構建了三元相變材料Ge2Sb2Te5的勢函數模型,實現了含7 200個原子的非晶體系的大規模分子動力學模擬,揭示了模型大小和原子個數對非晶局域結構的影響,并闡明了非晶構型中化學鍵和晶化過程中的微觀結構演化;Zhang 等[47]開發了適用于高性能并行計算的深度神經網絡勢函數方法DeePMD,實現了模擬體系超過1 億原子、模擬時長超過1 ns 的大規模分子動力學模擬,顯著加速了新材料設計與研發。
4 多尺度集成可視化的高通量自動計算和數據管理智能平臺ALKEMIE
2016 年,筆者團隊在國家重點研發計劃材料基因工程專項的支持下,基于Python 開源框架自主開發了一套多尺度集成可視化的高通量自動計算和數據管理智能平臺ALKEMIE,主要包含高通量自動工作流ALKEMIE Matter Studio(MS)、數據管理及材料數據庫ALKEMIE Database(DB)、基于機器學習的材料數據挖掘ALKEMIE Potential Mind(PM)三部分[14-15]。
4.1 高通量智能計算ALKEMIE-MS
ALKEMIE 基于AMDIV 設計理念,解決了材料高通量智能模擬過程中5 個核心問題:自動化計算(automation)、模塊化拓展(modular)、材料數據庫(database)、人工智能方法(intelligence)和可視化界面(visualization),可實現從建模、運行到數據分析,全程自動無人工干預。
ALKEMIE 中,多尺度集成的高通量自動計算可以通過不同模塊間以搭積木的方式實現自動耦合并完成計算。 不同模塊的連接方式如圖8 所示。 首先,由建模模塊控制輸入,通過多種建模方式將材料構型導入高通量預處理器,科學計算模塊控制任務的計算順序和糾錯(可進行電子尺度、原子尺度、分子尺度和介觀尺度的高通量自動流程)。 然后,服務器用來協調計算資源,配置遠程節點,實現本地與遠程服務器通信并提交任務,數據存儲系統負責保存整個流程中所有的元數據,將計算結果保存在不同類型的數據庫中。 最后,通過數據分析和人工智能進行數據挖掘。
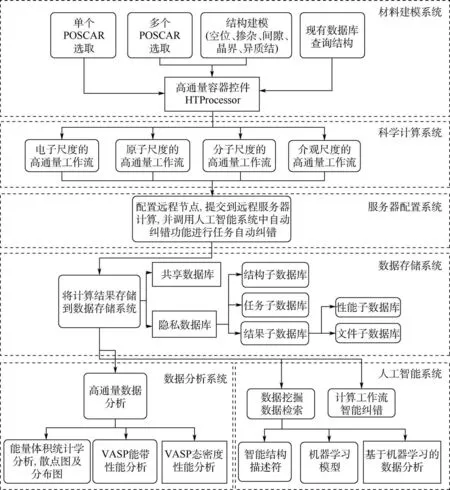
圖8 多尺度集成可視化的高通量自動計算和數據管理智能平臺ALKEMIE 計算模塊概況Fig.8 Overview of platform with multi-scale integration of visualized automatic high-throughput calculation and intelligent data management ALKEMIE
目前,該軟件可以實現超過104量級的高通量并發計算,包含第一性原理計算VASP、QE 和ABINIT,分子動力學模擬LAMMPS 和ASE,熱力學計算軟件Gibbs,動態蒙特卡羅模擬(KMC),相場相圖模擬OpenPhase 和OpenCalphd 等多尺度模擬軟件,可通過參數傳遞的方式實現跨尺度計算。 目前,該軟件系統已部署在9 家超算中心,包括4 家國家超算、4 家高校超算和1 家企業超算。軟件可移植性和可拓展性強,適用于對材料模擬掌握程度從初級到專業的所有材料研究人員,可提供材料建模、高通量智能計算、數據挖掘和人工智能一體化的新材料設計方案。
4.2 材料多類型數據庫ALKEMIE-DB
ALKEMIE-DB 材料多類型數據庫分為五大類:材料結構數據庫、工作流源數據庫、材料性能數據庫、機器學習描述符數據庫和論文數據庫。目前,收錄了超過64 萬條材料結構數據、296 條相圖數據、1 萬條聲子能帶數據、1 418 條贗勢數據和20 余萬條機器學習描述符數據。 通過ALKEMIE中JSON 格式的API 和國際通用的OPTIMADE 格式的API 實現數據高效查詢檢索,并通過Finder 控件實現結構的數據可視化,如圖9 所示[14]。

圖9 ALKEMIE-DB 材料結構數據庫[14]Fig.9 Material structure database of ALKEMIE-DB[14]
4.3 材料學機器學習應用框架ALKEMIE-PM
機器學習在材料學中通常被視為未知的黑盒模型,而材料研究人員相比于機器學習方法更關注材料性能、成分和工藝問題,因此,如何構建簡單通用的可視化機器學習框架至關重要。 ALKEMIE 通過抽象凝練高級API 及規范化和格式化機器學習的每個具體步驟開發了一套通用的可視化機器學習的流程。 Datasets 模塊給定了數據集及特征的輸入格式,Model 模塊定義了不同的機器學習算法,Evaluate 模塊給出了機器學習訓練過程的收斂情況及模型在測試集或模型在部署過程中的應用情況,Plotter 模塊中多種分析方法提供了將訓練過程及其結果可視化的功能,如圖10所示。

圖10 ALKEMIE-PM 機器學習框架Fig.10 Framework of ALKEMIE-PM machine learning
目前,筆者團隊基于ALKEMIE 可視化機器學習框架發展了一系列高效機器學習模型。Chen 等[48]通過團簇展開法和高通量第一性原理計算搜索了二維過渡金屬硫化物的單層和雙層無序摻雜結構,得到了穩定摻雜結構和能量之間的對應關系,通過機器學習挖掘了影響半導體-金屬轉變(SMT)的2 個關鍵特征,即范德華間隙內摻雜氧原子的濃度差和Mo-S/O 鍵角正切的平均值(tanθ);Peng 等[49]通過線性回歸方法構建了適用于MXene 材料單原子催化劑結構與氧還原反應(ORR)性能構效關系的組分描述符,基于簡單的元素性質可以高效快速預測材料的催化活性;Gan 等[50]通過高通量篩選和可視化機器學習框架,開發了2 個精度分別為90.90%和91.67%的機器學習模型預測層狀IV-V-VI 族半導體不同溫度下的最大熱電優值(ZTmax)和實現ZTmax所對應的最佳摻雜類型,并成功從840 個候選成分中篩選出數種具有優良潛力的熱電材料。
4.4 未來的發展方向
綜上所述,ALKEMIE 已經研發了集可視化高通量自動計算流程、材料多類型數據庫和人工智能方法于一體的新材料設計研發智能平臺,但是未來仍有亟須發展的新方向和新方法。
在高通量計算方面,開發從原子、分子、介觀到器件的跨尺度模擬方法是目前極具挑戰且具有廣闊應用前景的熱點問題。 Martin Karplus、Michael Levitt 和Arieh Warshel 三位科學家在分子領域憑借量子力學和分子動力學跨尺度模擬方法(QM/MM)獲得了2013 年諾貝爾化學獎。 而在周期性材料的研究方面,由于體系周期性邊界條件和原子局域環境的復雜性,使得跨尺度模擬的精度非常難以控制,發展高通量跨尺度高并發、自動糾錯及數據耦合方法,通過機器學習數據挖掘等算法進一步提升跨尺度模擬精度是未來的研究熱點之一。
材料數據庫方面,應該保持開源和共享的發展理念,基于FAIR 原則,構建包含材料計算和實驗元數據及中間數據的高效數據庫,發展數據規模更大、種類更豐富且具有航空特色的共享數據平臺,完善更加通用兼容的數據標準和共享標識均是未來重要的研究方向。
在機器學習領域,材料學中數據集的構建非常困難,因此,研發基于小數據集的高效機器學習模型訓練算法至關重要;由于機器學習模型的黑盒特性,探索可解釋的機器學習模型,闡明模型背后隱藏的物理意義,實現逆向材料成分和結構設計也是未來的熱門研究領域。
5 結束語
材料基因工程顛覆了傳統的“試錯-糾錯”材料研發模式,通過數據驅動的高通量方法和人工智能模型加速新材料的研發與設計。 本文系統總結了國內外知名的材料高通量計算框架、常用的針對多種材料體系的大型材料數據庫和機器學習方法,并概述了多尺度集成可視化的高通量自動計算和數據管理智能平臺ALKEMIE 的研究進展,提出了未來發展的研究方向,為實現按需逆向設計新材料提供參考。
猜你喜歡
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
華東師范大學學報(自然科學版)(2017年1期)2017-02-27 13:41:08
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
財經(2015年3期)2015-06-09 17:41:31
財經(2014年21期)2014-08-18 01:50:18
財經(2014年6期)2014-03-12 08:28:19
財經(2013年6期)2013-04-29 17:59:30
