磷酸摻雜型高溫質子交換膜燃料電池關鍵材料研究進展
2022-10-13 09:59:06相艷李文郭志斌張勁盧善富
北京航空航天大學學報 2022年9期
關鍵詞:結構
相艷 李文 郭志斌 張勁 盧善富
(1. 北京航空航天大學 空間與環境學院, 北京 100083; 2. 北京海得利茲新技術有限公司, 北京 100162)
燃料電池是一種將化學能直接轉換為電能的電化學能源轉換裝置,具有功率密度大、能量轉換效率高、環境污染小等眾多優點[1-3],可廣泛應用于便攜式電源、電動汽車等領域。 因此,發展燃料電池技術對解決能源與環境問題具有重要的社會意義。 其中,高溫質子交換膜燃料電池(HT-PEMFC)由于較高的工作溫度(130 ~220℃),使其具有較快的電極反應動力學、較高的雜質氣體(CO,SO2等)耐受性、簡單的水/熱管理系統及燃料來源廣泛等優點,受到廣泛關注與應用[4-8]。
1 高溫質子交換膜燃料電池技術
HT-PEMFC 的核心部件是基于磷酸(PA)摻雜的質子交換膜(HT-PEM)和催化層組裝的膜電極(MEA),這對電池的輸出性能和長期穩定性有直接影響[9-10]。 在PA 摻雜型HT-PEM 中,通常聚合物高分子鏈中主鏈骨架提供膜材料的力學性能,堿性/弱堿性基團提供PA 吸附位點。 PA 分子以結合和游離2 種形式存在于聚合物高分子鏈的網絡結構中,其中結合形式的PA 以強氫鍵作用“錨定”在聚合物高分子鏈周圍,而游離形式的PA(也稱“自由PA”)與聚合物分子相互作用較弱[11],主要在PA 分子間形成氫鍵網絡,氫鍵網絡是高溫質子傳輸的主要來源。 質子通過氫鍵網絡以Grotthuss 機制進行傳輸,即質子跳躍機制。 膜內的質子供體和質子受體之間可形成氫鍵,并互相連接構成氫鍵網絡,在該網絡中,質子沿著氫鍵在載體分子間移動,在相鄰分子間每完成一次跳躍,隨后都發生載體分子的重排,即氫鍵的形成和斷裂,這種不斷重排過程形成質子的傳輸通道。因此,較高的游離PA 摻雜能夠獲得較高的質子電導率[12]。 然而較高PA 摻雜水平會導致膜中大量的游離PA 分子填充在聚合物高分子鏈的周圍,減弱了分子鏈之間的范德華力,導致膜的尺寸穩定性大幅降低[13-14],在實際應用過程中面臨質子傳輸性能與機械強度難以協同兼顧的嚴峻問題。 對于催化層,在構建高溫膜電極(HT-MEA)時,在組裝壓力和催化層微結構毛細力作用下,PA 從電解質膜內遷移進入催化層中,并在電流驅動下在催化層中進行再分布,形成電化學反應三相界面。 然而,PA 在MEA 內的動態遷移過程使得PA 填充在催化層微孔結構中,造成反應氣體與催化劑的接觸受阻,增大了物質傳輸電阻[15];并且由于PA 及PA 解離的陰離子吸附在鉑基催化劑表面,降低了催化劑反應活性[16-17]。
近年來,人們對HT-PEMFC 存在的上述科學問題取得一定進展,但實現HT-PEMFC 保持優異的電化學性能、技術國產化及產品商業化發展仍然面臨著巨大挑戰。 人們希望通過廉價膜材料的選擇以降低使用成本,聚合物高分子結構設計以優化膜材料性能,大尺寸MEA 的一致性工藝與性能優化,HT-PEMFC 與甲醇重整器等耦合聯用以拓寬燃料氣體選擇范圍等途徑,有效解決高性能HT-PEMFC 的技術開發與實用化。
本文在梳理HT-PEMFC 關鍵科學問題的基礎上,著重針對北京航空航天大學(簡稱北航)相艷教授團隊近十年來在HT-PEMFC 關鍵材料的相關研究進展方面進行了詳細闡述。 如圖1 所示,主要包括:發展了3 代不同體系的HT-PEM 材料;調控了催化層微觀結構,實現其穩定化運行;制備了大尺寸MEA 并對其一致性工藝進行了深入研究等工作。

圖1 北航相艷教授團隊在高溫膜、催化層和膜電極與電堆的研究發展歷程Fig.1 Research and development history of Professor Xiang Yan’s team of Beihang University in high-temperature membrane,catalytic layer, membrane electrode and stack
2 高溫質子交換膜
在眾多高溫膜材料體系中,PA 摻雜型PEM由于在高溫低濕環境下具有較高的質子傳導率而被認為是最具發展應用前景的高溫聚合物膜材料[18-22]。 以早期研究中典型的PA 摻雜聚苯并咪唑(PA/PBI)為例,其骨架上的堿性咪唑基團在摻雜PA 后,咪唑基團與PA 之間形成連續氫鍵,增強了質子傳導[23]。 然而,由于PBI 材料合成工藝復雜,成本較高,限制了其大規模應用。 因此,開發更高效且低成本的膜材料對于HT-PEMFC 的大規模應用與發展尤為重要。
筆者研究團隊自2010 年前后開展高性能低成本的聚合物膜材料的研究工作,從膜基材的選擇、微觀結構的調控及質子導體的篩選3 個方面入手,發展了3 代新型HT-PEMs 材料(見圖2)。第1 代:聚乙烯吡咯烷酮(PVP)基HT-PEM。 PVP具有優異的溶解性及熱穩定性能,且具有獨特的分子單元結構,PVP 含有的吡咯烷酮環上的堿性N 和O 原子均可在酸性環境中被質子化,類似于PBI 上咪唑基團可以吸收PA,這為PVP 制備高性能PEM 提供了理論前提條件[24];第2 代:具有微觀相分離結構的HT-PEM。 無論是PBI 基或PVP基,其聚合物的傳導功能基團均在主鏈或者相連主鏈上,因此PA 摻雜膜面臨質子傳導率和拉伸強度之間的最佳平衡點的問題。 受商業化Nafion聚合物獨特的高分子鏈結構啟發:側鏈親水的離子團簇提供質子傳輸通道,憎水的主鏈形成膜的骨架。 從分子工程角度對高分子鏈進行源頭設計,改變高分子鏈上吸附PA 功能基團的位置及個數,實現PA 限域聚集分布,形成類似于Nafion膜的微相分離結構,在不損失質子傳導率的同時降低PA 對主鏈的塑化作用,兼顧優異的質子傳導率與拉伸強度。 第3 代:寬溫域PEM。 在前兩代PEM 的研究中,均使用PA 作為質子導體,傳質性能非常依賴PA 摻雜量,而PA 與聚合物基體的相互作用較弱,在電池運行過程中易流失,在室溫條件下難以啟動,造成性能的衰減。 如能開發非PA 型固態質子導體,可徹底解決PA 流失問題,并可將電池操作溫度拓寬至低溫實現低溫啟動,拓寬工作溫度范圍。 因此,筆者研究團隊基于質子導體開發了第3 代新型PEM,以期使用新型固態質子導體(如有機膦酸、核殼焦磷酸錫)代替PA 進行質子傳輸,解決PA 流失對電池帶來的負面影響,實現高性能HT-PEMFC 的開發與應用。本節將逐一詳細介紹3 代HT-PEM 的研究進展。
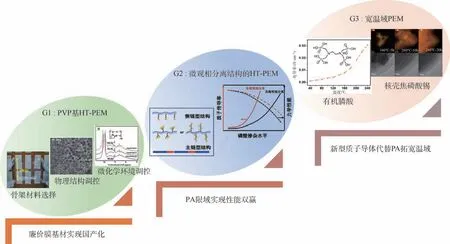
圖2 3 代不同HT-PEM 的設計理念與研究進展Fig.2 Design concepts and research progress of three generations of different HT-PEM
2.1 PVP 基HT-PEM
在PVP 基HT-PEM 中,PVP 的堿性N 雜環可用作PA 吸附位點,質子在PA 與N 雜環之間傳遞或PA 分子間的傳遞以實現質子傳輸[25-26]。 但由于PVP 脆性較大,一般不能單獨形成有用的膜材料。 因此,筆者研究團隊通過將PVP 與其他具有良好機械性能的骨架材料進行復合,并通過調控物理結構及微化學環境,制備出了高質子傳導率與優異機械性能的PVP 基HT-PEM 復合材料。
2.1.1 PVP 基PEM 骨架材料的選擇
聚偏氟乙烯(PVDF)作為一種化學穩定性優異,價格低廉的熱塑性材料,筆者研究團隊將其與PVP 混合制備了PVDF-PVP 復合膜[27],PVP 質量分數為80 %時,復合膜質子電導率在180℃可高達9 ×10-2S/cm。 然而由于PVDF 較低的玻璃化轉變溫度(Tg),當PVDF 含量為40%時復合膜的Tg僅為120℃,嚴重限制了高溫燃料電池的優勢。針對這一問題,本文采用Tg為230℃且化學穩定性良好的聚醚砜(PES)替代PVDF 與PVP 共混,制備了PES-PVP 高溫膜[26]。 當PVP 質量分數為80%時即PES-PVP 80%,復合膜的Tg仍可高達192℃(見圖3(a)),極大提高了復合膜應用溫度范圍。 此外,PVDF 與PVP 之間的氫鍵作用占據了部分PA 吸附位點,而PES 與PVP 之間沒有相互作用。 因此,PVP 的吡咯環中除N 原子外,羰基氧也被釋放出來吸附PA,使得PES-PVP 膜的PA吸附量(PA 摻雜水平為9.1)遠高于同樣80%PVP含量下的PVDF-PVP 復合膜(PA 摻雜水平為2.7),在180℃質子電導率達0. 21 S/cm (見圖3(b)),并獲得850 mW/cm2峰功率密度。 以上研究表明不同復合材料對PVP 基膜的物理化學性質有重要影響。 Ren 等[28]證實了與摻有脂肪族聚合物相比,芳族聚合物(如PES 和PSU 等)與PVP 制備的復合膜表現出更小的體積溶脹及更高的酸摻雜量,獲得高電導率的同時具有良好的機械性能,更適合應用于PEM。
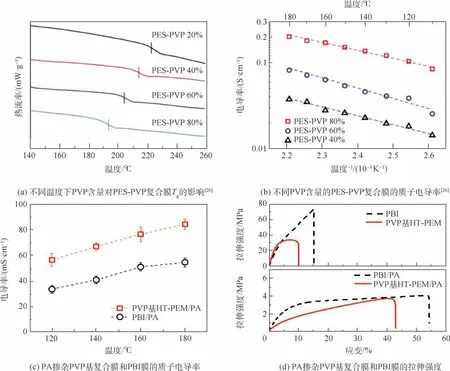
圖3 PES-PVP 膜的Tg 和質子傳導率及PVP 基膜和PBI 膜的性能對比Fig.3 Tg and proton conductivity of PES-PVP membrane and performance comparison between PVP-based membrane and PBI membrane
此外,筆者研究團隊還以HT-PEM 商業化應用為目標,成功使用流延法放大制備了小批量幅寬為40 cm 的PVP 基復合膜,實現了國產化應用。該膜中每個PVP 結構單元的吸PA 水平可達4.9,質子傳導率在180℃達85 mS/cm(見圖3(c)),優于同等條件下商業化PBI/PA 膜,摻雜PA 后的機械性能與PBI/PA 膜相當(見圖3(d))。
增加PVP 含量可有效提高膜的PA 摻雜水平,提高電導率,但也會嚴重影響膜的機械性能[26]。 有文獻表明,多孔聚四氟乙烯(PTFE)作為增強材料可顯著提高拉伸強度[29]。 筆者研究團隊在PES-PVP 膜的基礎上使用同時具備親疏水結構的Triton 表面活性劑作為PES-PVP 膜與PTFE 之間的界面,提高了二者之間的相容性。 摻雜PA 后,優化后的復合膜PES-PVP/PTFE-5(PTFE 的摻雜量為5%)(6. 50 MPa)相較PES-PVP膜(1.54 MPa)拉伸強度得到明顯提升,質子電導率在180℃達0.26 S/cm,并獲得607 mW/cm2的峰功率密度,且運行過程中開路電壓無明顯下降[30]。
2.1.2 物理結構與微化學環境調控
為了進一步提高質子傳導率,在不破壞聚合物主鏈結構的情況下對PEM 結構進行調控,提高局部自由PA 含量和自由PA 之間的連通性,同時也減弱PA 對主鏈的塑化作用,加強質子傳遞的同時保證膜具有良好的機械強度。 研究表明,在PEM 中引入多孔結構有利于負載大量自由PA[31]。 筆者研究團隊基于PES-PVP 復合膜,以單分散的SiO2固體球為硬模板制備了含亞微孔結構的PES-PVP/SiO2膜[32]。 獲得的多孔結構顯著提高了局部區域的自由PA 含量和自由PA 之間的連通性,在優化的孔結構下SiO2含量為50%的多孔膜(mp-50)質子傳導率在180 ℃可高達0.09 S/cm(見圖4 (a)), 峰功率密度為 454 mW/cm2(見圖4(b))。

圖4 以SiO2 為模板制備的PES-PVP 多孔膜及g-C3 N4 摻雜PES-PVP 復合膜的質子電導率和燃料電池性能Fig.4 Proton conductivity and fuel cell performance of PES-PVP porous membrane prepared with SiO2 as template and PES-PVP composite membrane doped with g-C3 N4
有機-無機共混作為提高PEM 尺寸穩定性及機械性能的方式之一,因制備方式簡單受到廣泛應用[12,33-34]。 筆者研究團隊將具有二維片層結構的石墨相碳氮化合物(g-C3N4)納米片(CN)引入PVP 基PEM 中[35],利用CN 中豐富的含氮功能基團(—NH 和C—N—C) 提高了PA 摻雜量(見圖4(c))。 此外,1H MAS NMR 結果表明,CN 能夠促進PA 以及PVP 上的質子解離,這歸因于CN的堿性(解離常數pKa=8.95)強于吡咯烷酮環堿性(pKa=7.6),更易與PA 形成氫鍵,優化了質子傳遞環境。 優化的復合膜P/CN-0.5/PA(P 代表PVP 基膜,CN 代表g-C3N4,0. 5 為CN 的含量為0.5%)在160℃的質子傳導率達104 mS/cm,拉伸強度為6. 0 MPa,分別比PES-PVP 膜提升了36%和60%,并獲得優異的恒壓放電穩定性(見圖4(d))。 除了物理共混,還通過化學交聯制備了PVP 與氯甲基聚砜的交聯膜[36],利用交聯生成的季銨基團增強了膜的PA 吸附能力,在160℃獲得了120 mS/cm 高質子傳導率。
在眾多無機材料中,磷鎢酸(HPW)作為最具吸引力的無機固體質子導體具有優異的穩定性及質子傳導性能,在室溫和100 % 濕度(RH)下獲得高達0.18 S/cm 電導率,被廣泛引入到聚合物基質中進行膜修飾或制備新型PEM[37-42]。 筆者研究團隊在前期工作中將HPW 負載到介孔二氧化硅MCM-41 中,并成功將其用作燃料電池的無機固體電解質,高效的微孔限域結構使得HPW納米團簇構筑了有效的質子傳輸途徑,最終獲得優異的H2-O2燃料電池性能及甲醇燃料電池性能[43]。 基于此,進一步探究了HPW 與PVP 膜基質的作用機制,PVP 吡咯環的N 原子可與HPW上的質子作用生成了質子化NR3H+,從而將HPW 錨定在聚合物網絡結構中,構建質子傳輸通道。 在60℃及全濕狀態下,質子電導率達到0.066 S/cm,與同樣測試條件下的Nafion 115 質子電導率相當[39]。 此后,本文將HPW 用于HT-PEMFC[44],1H NMR 結果表明摻有HPW 的復合膜比無HPW 摻雜膜表現出更高的可移動質子含量,顯示出更窄的線寬信號,表明該復合膜中聚合物鏈更易移動。 在160℃獲得1.44 ×10-1S/cm 高質子傳導率,單電池的峰值功率密度為416 mW/cm2。PA 作為一種中強酸在膜中難以完全解離,膜內質子濃度遠低于PA 濃度,使得PA 利用率較低。 綜上研究可知,可以通過復合其他無機材料優化膜內質子傳輸環境,提高PA 解離度,獲得更多可移動自由質子,進而提高質子傳導率。
2.2 微相分離結構HT-PEM
通過2.1 節研究中的物理增強、化學交聯等方式雖然很大程度可以改善膜材料的質子傳導性能與機械性能,但同時也會帶來一系列新的問題:纖維增強的復合膜吸收PA 后易出現增強纖維與膜基體剝離的問題;多孔結構的膜存在PA 流失速度快、穩定性差的問題。 因此,解決HT-PEM 目前所面臨的困境,必須從膜材料的化學結構出發,對聚合物高分子鏈進行源頭設計。 筆者研究團隊針對高分子鏈進行了精準設計,改變可吸附PA的功能基團的位置和個數(見圖5(a)),制備了一系列具有微相分離結構的新型膜材料,實現PEM 質子傳導率和機械性能雙贏(見圖5(b))。
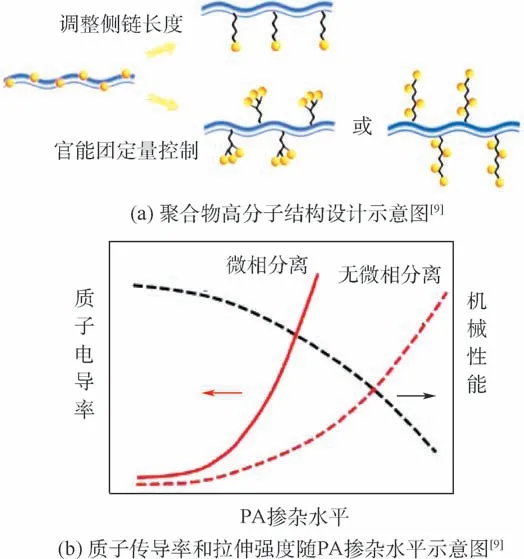
圖5 微相分離結構質子交換膜的設計與性能示意圖Fig.5 Schematic diagram of design and performance of PEM with microphase separation structure
通過調控聚苯醚聚合物膜(PPO-PIm-Cx)的末端咪唑官能化的側鏈長度調控膜的PA 吸收水平,在側鏈亞甲基個數為6 的PPO-PIm-C6 膜中,膜內形成明顯的微觀相分離結構,團簇尺寸約為5.56 nm,在160℃獲得質子傳導率0.042 S/cm,以及良好的拉伸強度12.1 MPa。 隨后,在另一項工作中探究了側鏈堿性基團數量對質子傳導率和機械性能的影響,將具有3 個叔胺基的2,4,6-三(二甲胺甲基)-苯酚(TDAP)接枝到PSU(TDAPPSU)上,與單叔胺接枝的PSU 膜(DMA-PSU)相比,1 個接枝位點上的胺基束明顯減少了體積膨脹。 接枝度75% 的TDAP-PSU 膜與接枝度99%的DMA-PSU 膜的PA 吸收量相當,但前者的機械強度卻是后者的2.2 倍。 基于TDAP-PSU 膜的單電池峰功率密度達到453 mW/cm2,且獲得優異的穩定性[45]。 此外,本文通過原子轉移自由基聚合將聚1-乙烯基咪唑(P-g-V)作為PA 摻雜位點接枝到聚砜骨架上,獲得系列具有不同側鏈長度和功能基團數量的聚合物材料[46],其中P-g-V-3.82/PA(每個接枝位點平均接枝3.82 個基團)復合膜在160℃的質子電導率為127 mS/cm,膜的拉伸強度保持為7.94 MPa(見圖6(a)),遠高于相同測試條件下PBI/PA 膜。 優異的性能歸因于P-g-V-3.82膜的功能性側鏈自組裝形成微相分離結構,如圖6(d)所示的原子力顯微鏡(AFM)圖像顯示膜內出現側鏈的親水微相(暗區)和聚合物主鏈的疏水微相(明亮區),進而實現了質子快速傳輸并維持了良好的機械性能。
然而,側鏈型HT-PEMs 的熱穩定性和化學穩定性仍有待提高,一般而言,類似PBI 主鏈型聚合物的熱穩定性要優于側鏈結構的聚合物。 若結合主鏈型聚合物優異的熱穩定性及側鏈型微相分離結構,設計一類摻雜PA 后可以形成微相分離結構的主鏈型聚合物,則可以更好地兼顧質子傳導性、拉伸強度以及熱穩定性的問題。 因此,本文研究團隊制備了基于剛性疏水芳基骨架和哌啶基團的聚亞芳基哌啶(PPT)[47]。 通過AFM(見圖6(d))和小角X 射線散射(SAXS)(見圖6(b))測試發現,OPBI 膜在摻雜PA 前后均未形成微相分離結構。而PPT/PA 膜在AFM 圖像中出現明顯的微相分離結構區域(見圖6(d)),其中暗區和亮區分別代表來自哌啶環基的親水微相和來自聚合物骨架的疏水微相,且SAXS 數據顯示在PPT 膜內成功形成了PA 聚集的離子團簇(見圖6(b))。 良好的微相分離結構使得PA 更易聚集在哌啶環周圍,與憎水的苯環分離,降低了PA 對分子鏈的塑化作用,獲得了12 MPa 的優異拉伸強度,在180℃無外部加濕條件下顯示出96. 0 mS/cm 的高質子電導率(見圖6(a)),且在質子傳導率循環穩定性測試中PPT/PA 膜始終優于OPBI/PA 膜(見圖6(b))。 在180 ℃基于PPT/PA 膜的H2-O2燃料電池的最高輸出功率高達1 220.2 mW/cm2,是同等測試條件下OPBI/PA 膜的單電池輸出性能的1.85 倍(660.8 mW/cm2),且在1 600 h 的耐久性測試中保持了優異的輸出穩定性(見圖6(c))。

圖6 P-g-V-3.82/PA 膜、PPT/PA 膜及OPBI/PA 膜的微相分離結構及性能對比Fig.6 Comparison of microphase separation structure and performance of P-g-V-3.82/PA membrane, PPT/PA membrane and OPBI/PA membrane
2.3 寬溫域PEM
PA 摻雜PEM 的質子傳遞性與膜內PA 摻雜量有直接關系,然而小分子PA 易受到各種因素影響而流失[48-49],導致無法室溫啟動等問題[18]。盡管前人已做了很多努力,筆者研究團隊也已發展了2 代新型的HT-PEM,但對于PA 流失問題仍沒有徹底解決。 因此,筆者研究團隊從質子導體角度出發,擬發展新型質子導體代替PA 進行質子傳輸,進一步拓寬PEMFC 的操作溫度范圍:拓寬低溫區實現電池的室溫啟動,拓寬高溫區可將電池與甲醇重整器等進行耦合聯用。
研究證明有機膦酸(OPA)是一類良好的有機質子導體[50],較大的分子尺寸使其分子動態遷移能力低于PA,理論上不易流失。 已有研究表明,烷基或苯基取代OPA 在高溫無水條件下可獲得10-2~10-1S/cm 的高電導率[51-52]。 因此,將OPA 作為質子導體用于制備新型HT-PEM,有望解決PA 摻雜型HT-PEMFC 面臨的困境。 為此,筆者研究團隊探索了不同OPA 在HT-PEM 中的質子傳輸行為與機制。 將乙二胺四亞甲基膦酸(EDTMPA)引入PVP 膜基質中構建了新型PEM,EDTMPA 和PA 雙質子導體協同改善了單位體積膜內的有效質子濃度及復合膜質子擴散系數,使得該膜在180℃無水條件下質子電導率可達0.065 S/cm,約為僅含有單一PA 質子導體膜的1.8 倍,并獲得716 mW/cm2高峰功率密度及優異的輸出穩定性。 此外,EDTMPA 對PA 的錨定作用使得復合膜在80℃同樣獲得了優異的電導率(0.025 S/cm)及326 mW/cm2峰功率密度,這有望實現電池的低溫啟動。 此外,還發現氨基三亞甲基膦酸質子導體(ATMP)與OPBI 膜有明顯交聯作用,這區別于傳統僅有交聯作用的交聯劑。因此,該現象啟發本文將兼具質子傳導功能和交聯作用的ATMP 引入OPBI 膜中制備交聯膜,在較低 PA 摻 雜 量 下(156%) 在 80℃獲 得 了0.033 S/cm和在160℃0. 112 S/cm 的高質子傳導率,單電池分別在80℃達到了0.268 W/cm2和在160℃0.98 W/cm2的高峰功率密度。 除OPA質子導體之外,有研究發現,核殼焦磷酸錫(SnP2O7)在200℃以上也具有良好的質子電導率[53-55]。 筆者研究團隊對核殼SnP2O7在200℃及以上更高溫度的質子傳導機制進行探究發現,核殼SnP2O7優異的質子電導率主要來源于最外層的凝膠層,該凝膠層主要成分是焦磷酸,260℃無水條件下可獲得0.084 S/cm 的高質子傳導率,高溫電導率可穩定170 h 左右,在200℃以上的高溫條件下極具質子導體應用潛力。
綜上,筆者研究團隊共開發了3 代PEM,首先開發了低成本PVP 膜基材,探究了與其他骨架材料形成復合膜的性能,并調控了PVP 基PEM的物理結構和微化學環境以改善膜的質子傳導率與機械性能的平衡問題,并實現了PES-PVP 復合膜國產化批量制備;接著進一步通過高分子鏈設計制備了具有微相分離結構的PEM,將吸附PA的側鏈功能基團與提供機械性能的主鏈分離開來,實現質子傳導率與拉伸強度的雙贏;以上2 種策略均是基于開發新型膜材料且均使用PA 作為質子導體,而PA 帶來的一系列問題仍不可避免,因此,非PA 型質子導體的開發對于PEMFC 的實際應用至關重要,我們探究了新型質子導體用于PEM,并對其在寬溫域的傳導機制進行研究,進一步實現PEMFC 的實用化發展。
3 催化層
在PA 摻雜PEM 中,PA 受各種因素在MEA內進行分布與遷移[9,56-59]。 如圖7 所示,在電池運行過程中,膜內PA 受到電流驅動,PA 水解的H2PO-4從膜內向陽極遷移,與陽極側的氫離子反應形成PA,提高了陽極PA 含量。 隨后,由于濃度梯度差使得PA 擴散到陰極,與陰極產物水結合形成H2PO-4,此循環過程促進了PA 在催化層中的再分布,導致催化層出現“酸淹”現象,大量PA 以及PA 解離的負離子占據鉑催化劑的活性位點,使得催化層內的物質傳輸阻力增大。 因此,筆者研究團隊擬通過從催化層體相和界面等角度對PA 在MEA 中的分布進行適當調控,以期緩解PA 對MEA 造成的不利影響,提高燃料電池性能。
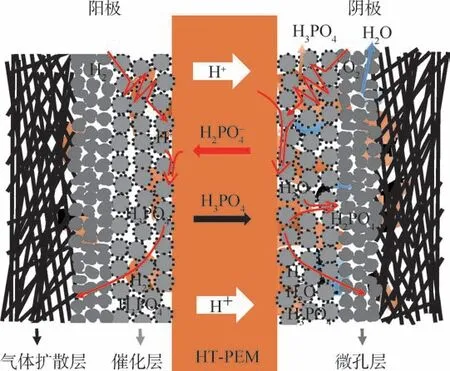
圖7 磷酸在膜電極中的分布與遷移示意圖[9]Fig.7 Distribution and migration of phosphoric acid in membrane electrode[9]
在催化層制備過程中,催化層的結構受到催化劑墨水中使用的溶劑沸點,黏結劑含量及制備方法等影響,導致在催化層中形成不同尺寸的裂紋[60-62]。 較大的裂紋會降低催化層的毛細力作用,使PA 快速遷移進入催化層內部,形成“酸淹”[63]。 Halter 等[64]發現PA 充填裂紋的毛細壓力隨著裂紋寬度的減小而增大,裂紋的連通性和可及性對PA 的保留能力起主要作用。 而催化層內的PA 壓力等物理特性易受到制備方法的影響。 因此,筆者研究團隊研究了超聲噴涂、絲網印刷、刮涂3 種方式構建的具有不同微觀結構的催化層(見圖8(a)),探究了制備方法對催化層微觀結構的影響[15]。 結果表明,超聲噴涂制備的催化層表面平坦,裂紋少且互相不連通,具有良好的二級孔分布,促進了催化層內PA 的均勻分布,構建了更多電化學反應三相界面;而絲網印刷和刮涂制備的催化層表面分別顯示出20 ~30 μm 和10 ~15 μm 的相互連通的明顯裂紋,表現出較發達的二級孔結構(見圖8(b))。 基于超聲噴涂制備的催化層HT-PEMFC 的峰功率密度高于其他2種方式制備的催化層(見圖8(b)),且保持了優異的穩定性。 因此,減少催化層裂紋的生成,降低裂紋程度,能夠有效避免催化層內PA 的不均勻分布,降低極化過程中的物質傳輸損失。

圖8 改善催化層裂紋對膜電極性能的影響Fig.8 Effect of improving catalytic layer cracks on membrane electrode performance
除了制備方法的優化,本文利用具有強韌性和高長徑比的一維柔性碳納米管[65-66](CNT)進一步消除溶劑蒸發過程中催化劑團聚體間的拉伸應力(見圖8(c)),達到降低催化層裂紋數量和寬度的目的[67]。 同時,CNT 高比表面積也提高了催化層內的二級孔含量,促進PA 在催化層內的均勻分布,致使電化學反應動力學得到提升,物質傳輸阻力降低。 當催化層中加入質量分數為2.0% 的CNT 時,電池峰值功率密度為673 mW/cm2,比不含CNT 的電池高1.5 倍,且在多次啟停過程中表現出良好的穩定性。
盡管加入一維CNT 材料可以抑制催化層內裂紋的生成及增加催化層二級孔含量,達到降低物質傳輸阻力和提高Pt 利用率的目的。 然而催化層的二級孔孔徑范圍跨度較大且大小不一,影響了催化層內PA 含量和分布,并且催化層內相互貫通的孔結構增加了PA 流失的風險。 因此,為了降低靠近膜側催化層的PA 含量,筆者研究團隊構建了具有更致密孔徑結構的界面層,在催化層與PEM 之間構建SiO2界面(見圖9(a)),利用納米SiO2較小粒徑(10 ~20 nm)、與PA 作用力強以及能與PA 在高溫反應的特性,有效地降低了催化層中PA 的初始含量,減緩了PA 向催化層的遷移速率,提高了HT-PEMFC 的輸出功率和穩定性。 基于SiO2界面層組裝的HT-MEA 即使在30℃,0. 3 A/cm2下也能夠穩定運行超過280 min(見圖9(b)),在30 K 圈電勢循環的加速老化測試過程中,其峰功率密度僅衰減15. 4%(見圖9(c)),遠低于沒有SiO2界面層組裝的高溫MEA(56.6%)。 通過對老化后催化層3D 結構重建分析,由于PA 向催化層遷移量降低,催化劑團聚體尺寸僅發生輕微的變化(見圖9(d)),合理的致密界面層有效改善了MEA 的輸出性能及穩定性。

圖9 SiO2 界面層對MEA 性能的影響Fig.9 Influence of SiO2 interface layer on MEA performance
4 高溫膜電極和燃料電池堆
隨著PVP 基復合膜的批量制備工藝逐漸成熟,本文將其與Pt/C 催化劑電極組裝成不同尺寸的MEA,進一步考察了其在HT-PEMFC 中的實際應用。 分別制備了25,200,400 cm2的大尺寸MEA。圖10(a)展示了基于MEA 尺寸為200 cm2的PVP 基PEM/PA 燃料電池在H2-O2環境下的極化曲線,電池表現出1.01 V 高開路電壓,表明膜極低的氣體滲透率,且展示出優異的輸出性能,在0.6 V 下的輸出電流密度為0.62 A/cm2。 隨后,在H2-空氣環境下,由于空氣中的氧氣分壓降低導致電池的開路電壓略微降低至0.98 V,0.6 V下的輸出電流密度為0. 32 A/cm2。 為了評價該MEA 的商業化水平,在相同測試條件下對美國Advent 公司和丹麥電力系統公司制備的基于PBI/PA 的MEA 性能進行測試,2 種基于PBI/PA膜的MEA 電池的開路電壓與本研究的PVP 基PEM/PA 的電池基本一致(0.98 V),0.6 V 下的輸出電流密度分別為0.39 A/cm2及0.25 A/cm2,而基于PVP 基PEM/PA 膜的MEA 性能處于上述2 種商業化MEA 之間,表明本研究的大尺寸MEA具有極大的商業化潛力(見圖10(b))。 MEA 的運行穩定性對于燃料電池的實際應用至關重要。在150℃,0.2 A/cm2電流密度下對PVP 基PEM/PA膜燃料電池進行穩定性測試,如圖10(c)所示,電池在長達3 000 h 的運行時間內表現出優異的穩定性,電壓衰減速率僅為9. 7 μV/h,與商業化PBI/PA 膜在同等放電條件下性能相當。 基于單電池良好的輸出性能及穩定性,將其組裝成燃料電池電堆(MEA 面積為200 cm2)進行測試。 采用多蛇形流場,并對其進行優化,獲得更高輸出性能以及更均勻的電流密度分布(見圖10(e))。 如圖10(d)所示,具有3 片膜電極的電堆具有高的開路電壓,證明其具有良好的密封性能。 在穩定性測試中,分別在電堆組裝完成的第1 ~17 天和擱置100 余天后,對電堆進行從室溫升至工作溫度的啟停、輸出性能測試及停機氮氣吹掃等工序來測試其穩定性。 發現在整個測試時長內,電堆的開路電壓基本保持在3 V,且輸出性能幾乎維持在穩定水平。 在第121 天和第162 天的測試中,輸出電流密度較初始性能略有降低,這可能是催化層吸潮所致。 此外,本文還繼續組裝了20 片大尺寸MEA 的長電堆,開路電壓達到20 V,150℃下12 V 下的輸出電流達到45 A,且獲得了高達1.15 kW 的高峰功率密度(見圖10(f)),初步實現了PVP 基PEM/PA 燃料電池千瓦級輸出性能的目標,并且到目前為止,本文嘗試將其與甲醇重整器系統耦合(見圖10(g)),其部分成果已經在北京海得利茲新技術有限公司實現產業化轉化。 以上研究為HT-PEMFC 關鍵材料以及燃料電池的國產化提供了重要的研究基礎。

圖10 基于PVP 基PEM 組裝的不同尺寸的膜電極性能及部分器件實物照片Fig.10 Performance investigation of membrane electrodes with different sizes assembled based on PVP-based PEM and photos of some devices
5 結 論
HT-PEMFC 具有較強的雜質氣體耐受性、廣泛的燃料選擇性及可實現高效熱電聯動等諸多優勢,且其發展迅速,目前成為燃料電池未來的主要發展方向之一。 本文基于HT-PEMFC 的關鍵部件進行了廣泛深入的探究。
1) 對聚合物膜材料進行開發和探究,分別發展了PVP 基PEM、微相分離結構PEM 及寬溫域PEM。 PVP 基PEM 得益于PVP 吡咯烷酮環上的官能基團對PA 的吸附作用,并通過物理化學調控顯著提升了膜的酸吸收量及質子傳導率,并且初步實現了PVP 基復合膜的批量制備及國產化應用;進一步通過對聚合物高分子鏈進行精細設計,構筑了具有微觀相分離結構的新型高溫膜材料,使得PA 限域聚集分布,保證質子傳導率的同時降低PA 對主鏈的塑化作用,實現質子傳導率與拉伸強度雙贏;此外,PA 摻雜PEMFC 的PA 流失問題嚴重影響其實際應用,通過發展基于新型非PA 質子導體的寬溫域PEM,利用有機膦酸或核殼焦磷酸錫質子導體代替PA 進行傳質,有望緩解PA 流失,降低電池啟動溫度,縮短啟動時間,另一方面新型質子導體使得電池工作溫度提高,使其與甲醇重整器耦合,提升熱利用率和能量轉化效率。 因此,兼顧質子傳導率與拉伸強度的低成本且具有寬工作溫域的電解質膜材料是后續研究的重點方向之一。
2) 對于催化層,基于PA 在MEA 內的遷移和分布,對催化層的微觀結構進行系列調控,包括優化催化層的制備方法以及引入納米材料CNT 進一步提升性能,還通過在三相界面構筑更致密的SiO2層,調控PA 分布,緩解PA 流失。 然而,PA在催化層內的遷移是一個動態過程,目前缺乏更多的原位測試手段,后續研究可以此為方向,進一步明晰其動態遷移過程,為催化層設計提供依據。
3) 為了實現商業化應用,對基于PVP 復合膜的大尺寸MEA 的制備及一致性工藝進行探究,并成功組裝了千瓦級HT-PEMFC 電堆,展示出優異的電池穩定性。 綜上系列工作為HT-PEMFC 關鍵組成部件的發展提供了切實的參考依據,更高性能膜材料的開發,以及MEA 的商業化發展仍是PA 摻雜PEMFC 未來的重點研究方向之一。
此外,HT-PEMFC 普遍使用的質子導體PA對催化劑的毒化作用也是限制HT-PEMFC 商業化和大規模應用的重要影響因素。 因此,開發低成本、高活性的抗PA 催化劑也是提高HT-PEMFC 穩定運行的重要方向之一。 目前研究表明,通過電子結構調控或物理阻隔等方式可以有效改善催化劑單電極活性,但實現其在HT-PEMFC 中的高效應用依舊面臨挑戰。 在后續研究中進一步發展可以表征PA 對催化劑毒化機制的原位表征手段,對于深入指導催化劑的理性設計及開發更有效的抗PA 毒化策略具有重要的科學意義。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
