銅箔集流體表面聚苯胺復合改性研究
2022-10-21 11:46:16付紅霞肖仁貴張云燕
電子元件與材料 2022年9期
付紅霞,肖仁貴,張云燕,廖 霞,杜 鑫
(貴州大學 化學與化工學院,貴州 貴陽 550000)
鋰離子電池(LIB)由于循環穩定、能量密度高[1],被廣泛應用于消費電子、動力電池及儲能等領域[2],同時由于電動汽車的快速發展對鋰離子電池的安全性、穩定性和能量密度提出了更高的要求[3]。LIB 主要由正極、隔膜、負極、集流體四個部分組成[4]。集流體是LIBs 的主要組件之一,用來維持電極和外部電路之間的電力傳輸,其幾何形狀和表面化學分子結構將影響電子轉移速率[5],從而影響電池的電化學性能。
銅箔通常用于鋰離子電池的負極集流體,作為電極材料、粘合劑和導電材料的載體[6]。但是由于銅箔表面比較光滑平整,接觸面積不夠大,且銅箔為金屬材料,而電池一般為非金屬材料,使得兩種物質接觸相容性差,界面接觸電阻較大,導致電池充放電性能有所降低。所以,銅箔集流體的改性成為提高鋰離子電池性能的有效方法之一[7-11]。Wang 等在銅箔集流體上制備了親鋰3D ZnO 納米棒陣列,降低了局部電流密度,抑制了鋰樹枝晶的生長,在電流密度為5 mA·cm-2下循環200 次后,鋰電池容量保持率可達到93%[12]。Jiang 等在銅箔表面采用LPCVD 技術制備了石墨烯薄膜[7],石墨烯表面改性可增加銅箔與電極材料的附著力,提高電池的循環穩定性。Wang 等使用簡單的滴涂法在鋰表面形成超分子PEO-UPY 共聚物鋰陽極保護層,該共聚物作為一種穩定的人工固態電解質界面,可進一步減少銅箔集流體與鋰的反應,形成致密的保護層,從而弱化鋰枝晶的形成,提高鋰金屬陽極的循環穩定性[13]。Li 等設計了一種帶有聚合物接枝的表面改性銅箔集流體,此集流體可以顯著改善由平面鋰金屬鍍層導致的鋰電化學沉積[14]。這些研究工作主要從電池的安全性與循環穩定性方面做了進一步改善,但鮮有通過銅箔集流體改性既能提高電池循環性能,又能有效提高電池容量的報道。
聚苯胺最早在1834 年被發現[15],但它的導電特性直到1960 年才得以研究[16]。苯胺聚合通常采用電化學方法: 恒定電流法[17]、恒定電壓法、循環伏安法[18]。在恒定電流下聚合得到的聚苯胺的粘附力較弱[19]。恒壓法能得到均勻的聚合物膜,膜的粘附能力強[20-21],通過電壓及電解質類型等實驗條件,可控制其導電性[22]。苯胺的陽極氧化通常是在金、石墨、玻璃碳和不銹鋼等不活潑的電極材料上進行[16],當活潑金屬材料如鐵、銅等用作聚苯胺聚合陽極材料時,金屬在苯胺電化學聚合過程中會發生陽極溶解,金屬電極的陽極溶解對聚苯胺的形貌、結構及性能產生重要影響[23]。因此,進一步研究在銅箔表面電化學制備聚苯胺工藝,探討聚苯胺形貌結構與電化學聚合工藝參數之間的關系,以及聚苯胺形貌結構與銅箔集流體電化學性能之間的關系顯得非常重要。
本文在前期恒流法制備復合銅箔集流體的基礎上[17],通過恒壓法在銅箔表面電化學制備聚苯胺改性集流體,探討電壓對聚苯胺形貌與結構的影響規律,以及改性銅箔集流體對電池容量及循環性能的影響。
1 實驗
1.1 實驗材料
高純電解銅箔(純度≥99.5%,8 μm);苯胺(阿拉丁);H2SO4(98.3%,重慶創東化工(集團)有限公司);商用Li4Ti5O12活性材料(深圳科景星科技有限公司);鋰片(深圳碧源電子科技有限公司);隔膜(Celgard 2400)。
1.2 樣品表征
采用Nicolet-iS5 型(Thermo Fisher Scientific)傅里葉變換紅外光譜儀對電化學聚合產物進行官能團測試;采用UV-2700i 紫外光譜(Shimadzu)進行分子結構分析;采用Zeiss Sigma 掃描電鏡表征復合材料表面形貌;循環伏安曲線(CV)和電化學阻抗譜(EIS)由VersaSTAT 3 電化學工作站(Princeton Applied Research)獲得。
1.3 制備材料
1.3.1 聚苯胺的電化學聚合
聚苯胺的電化學聚合過程示意圖見圖1。在1 L 燒杯中配置0.05 mol/L 的H2SO4溶液和0.2 mol/L 的苯胺混合溶液構成電解液,用氮氣驅氧15 min。銅箔為陽極,石墨板為負極,進行恒壓法電化學聚合。電化學聚合后,用去離子水清洗銅箔表面,烘干備用。
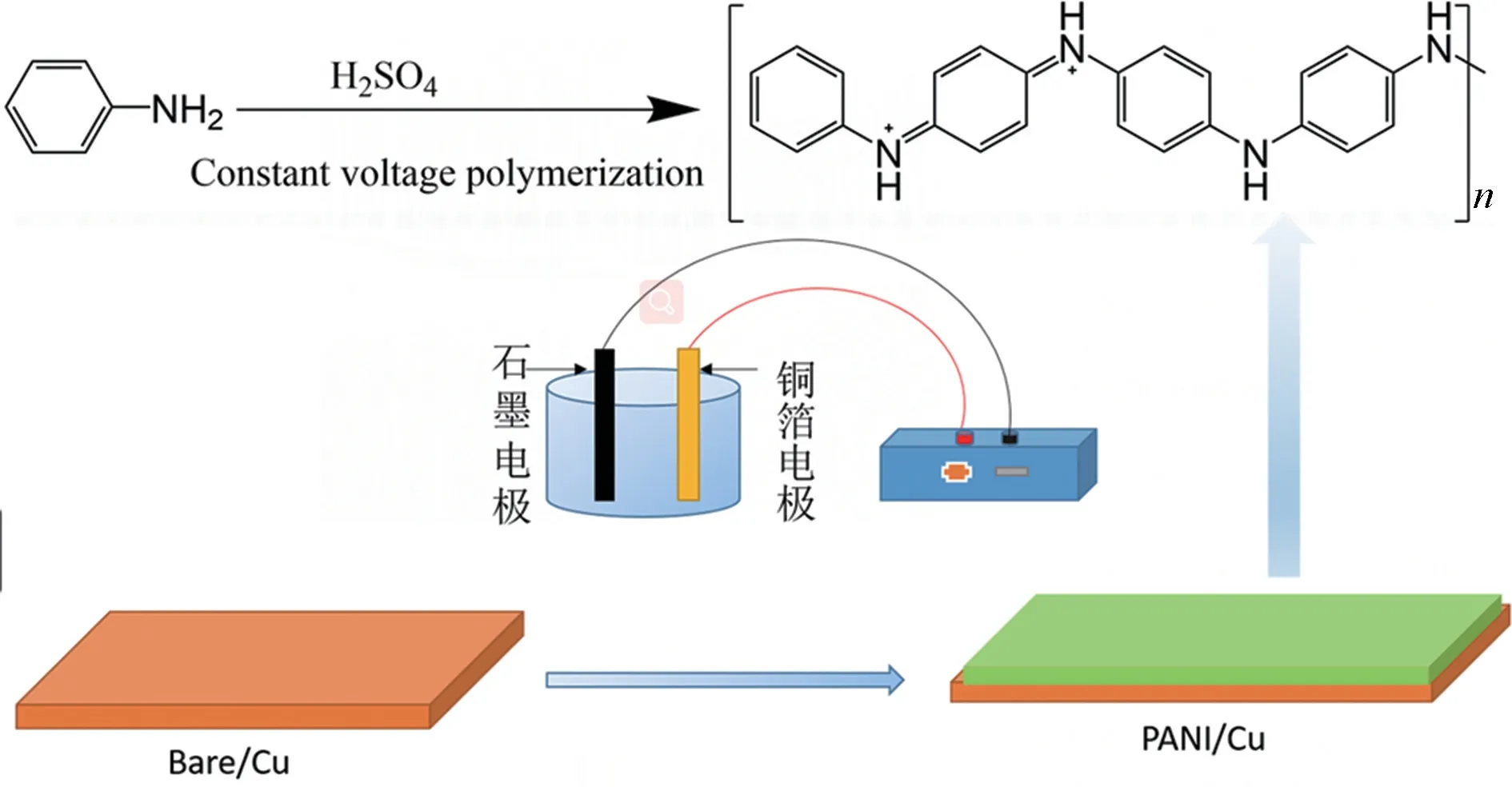
圖1 恒電壓聚合法制備復合銅箔集流體示意圖Fig.1 Fluid collection diagram of composite copper foil prepared by constant voltage polymerization
1.3.2 電池組裝
分別用LiTi5O12(LTO)為活性材料,乙炔黑為導電劑,聚偏氟乙烯(PVDF)為粘結劑,N-甲基吡咯烷酮(NMP)為溶劑,將LTO、乙炔黑及PVDF 按照質量比8 ∶1 ∶1 均勻混合,制成一定黏度的漿料,并均勻涂覆在PANI/Cu 與Bare/Cu 集流體表面,在120 ℃下揮發NMP 溶劑得到LTO 電極。分別用空白銅箔(Bare/Cu)與復合銅箔(PANI/Cu)作集流體,LTO 為負極,LiPF6(溶劑體積比EC ∶DMC ∶DEC=1 ∶1 ∶1)作電解液,組裝CR2032 紐扣電池,并使用藍電系統(CT2001A,武漢)進行恒流充放電測試(1.0~2.5 V Li+/Li)。
2 結果與討論
2.1 電壓對聚苯胺膜層形貌結構的影響
在0.2 mol/L 苯胺單體和0.05 mol/L 硫酸溶液組成的電解質溶液中,常溫恒壓聚合10 min,在不同電壓下聚合獲得的銅箔表面聚苯胺膜層微觀形貌見圖2。

圖2 不同電壓下聚合聚苯胺表面掃描電鏡圖。(a)Bare/Cu;(b)1.1 V;(c)1.2 V;(d)1.3 V;(e)1.4 V;(f)1.5 VFig.2 SEM images of polyaniline surface at different voltages.(a)Bare/Cu;(b)1.1 V;(c)1.2 V;(d)1.3 V;(e)1.4 V;(f)1.5 V
由圖2(a)可知,Bare/Cu 表面因為受到電解銅箔生產工藝的影響,導致其表面出現不規整的條紋狀。由圖2(b)所示,電壓為1.1 V 時聚苯胺顆粒分布不均勻,銅箔的部分表面并未形成均勻的聚苯胺顆粒層,這是因為電壓小,導致電流密度小,電聚合反應速度慢,聚苯胺顆粒在銅箔表面覆蓋不完整。電壓為1.2 V 與1.3 V 時,如圖2(c)與圖2(d)所示,銅箔表面均有孔洞。而當電壓為1.4 V 時,從圖2(e)中可觀察到均勻致密的聚苯胺顆粒層。隨著電壓增大至1.5 V 時,電聚合反應的速度增快,出現了聚苯胺顆粒不斷地堆疊聚集,出現了聚苯胺膜層堆積不均勻的狀況,結果如圖2(f)所示。因此后續表征及應用以電壓為1.4 V條件下制備的復合銅箔集流體為樣本。
2.2 電壓對聚苯胺膜層厚度的影響
采用萬分尺測厚儀測得不同電壓情況下所制備的聚苯胺膜層的厚度,測定結果如圖3 所示。通過圖3可知,隨著電壓的增大,聚苯胺生長的厚度也隨之變厚,但是當電壓達到1.3 V 以后,聚苯胺生長的厚度達到一個較為穩定的值,聚苯胺膜層的極限厚度可能與恒壓下的極限電流有關系,隨著電流達到一定值,聚苯胺的顆粒生長達到極限,而且聚苯胺難以在聚苯胺的表面繼續生長,所以會出現聚苯胺膜層厚度不再隨著電壓增加而增加的現象。
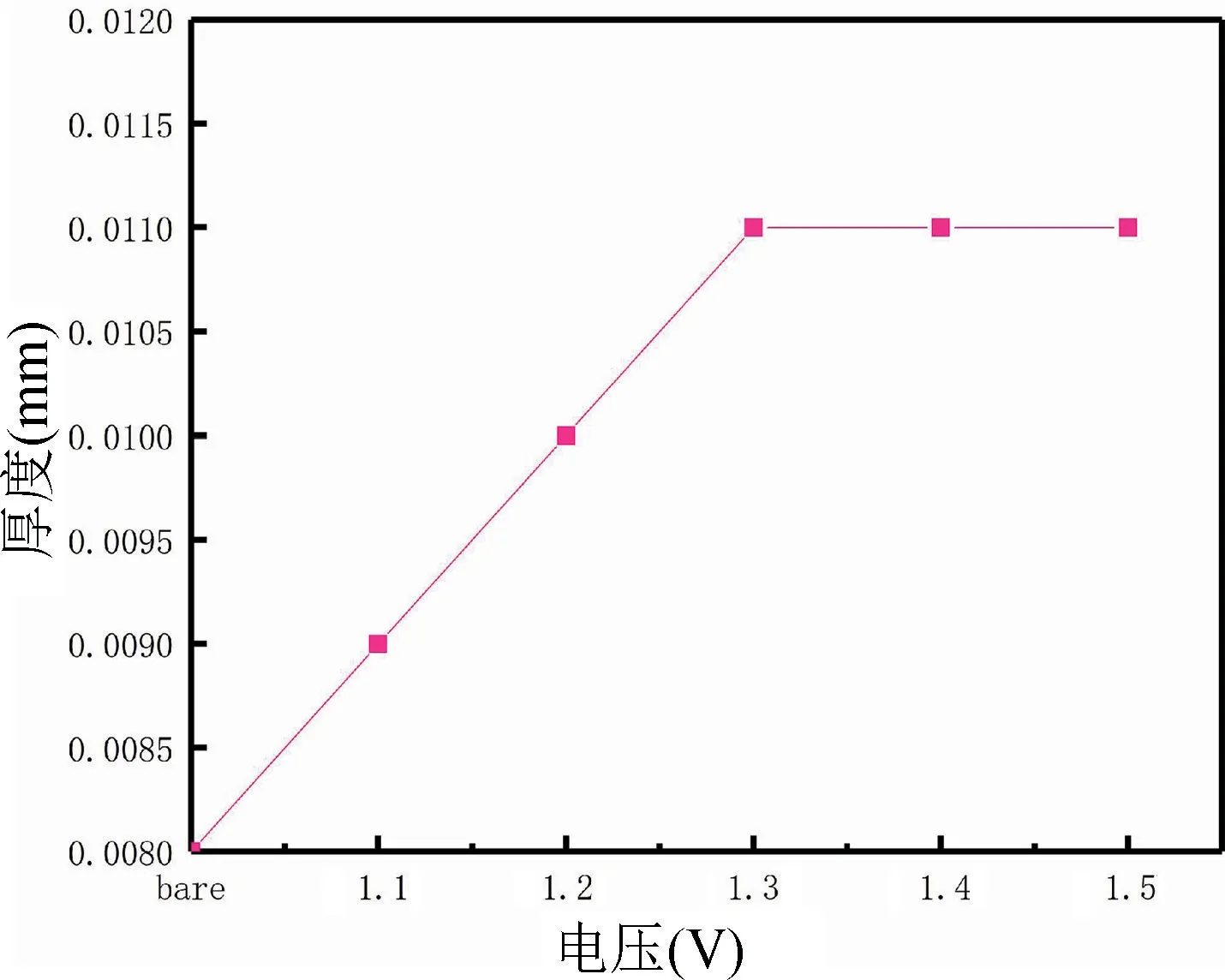
圖3 聚苯胺生長厚度隨著電壓的變化Fig.3 Variation of polyaniline growth thickness with voltage
2.3 時間對聚苯胺膜層均勻性的影響
在1.4 V 電壓下,由0.2 mol/L 苯胺與0.05 mol/L 的硫酸配成的混合電解質中,常溫條件下,分別電聚合1,5,10 和15 min,圖4 為聚合不同時間下聚苯胺膜層的表面形貌結構。
由圖4(a)可知,恒電壓聚合1 min 后聚苯胺膜層由粒狀結構構成,因為電聚合的時間較短,導致聚苯胺顆粒沒有完全覆蓋銅箔表面,得到厚度不均勻的聚苯胺膜層。由圖4(b)所示,恒電壓聚合5 min 后,聚苯胺顆粒分布相對于1 min 更均勻,但是還是有一些空隙。由圖4(c)所示,恒電壓聚合10 min 后,得到均勻堆積的PANI 顆粒,形成均勻致密并且平坦整齊的聚苯胺膜層。再繼續聚合到15 min 后,如圖4(d),聚苯胺在10 min 的基礎上繼續生長,可能是由于聚苯胺晶粒的生長導致聚苯胺相互擠壓并且產生裂痕。因此,在1.4 V 電壓下生長10 min 聚苯胺的表面形貌較為理想。
2.4 聚苯胺膜層的紅外光譜表征圖
圖5 為電化學聚合生成聚苯胺膜層的紅外光譜圖。圖5 表明,醌環(Q)和苯環(B)的特征吸收峰在1561 cm-1和1492 cm-1處,分別為氧化還原態聚苯胺的特征峰[24],該結構為導電態聚苯胺。由于特征吸收峰相對強度IQ/IB=I1561/I1492越接近1,聚苯胺電導率就越好。特征吸收峰出現在1117 cm-1是聚苯胺鹽結構,與—N=Q=N—伸縮振動相對應,而這個強寬帶與聚苯胺的高度電子離域和高電導率有關[25]。除此之外,苯環中C-H 的特征吸收峰在751 cm-1處[26]。通過以上分析可知,電化學聚合生成的聚苯胺以導電態形式存在于銅箔表面。
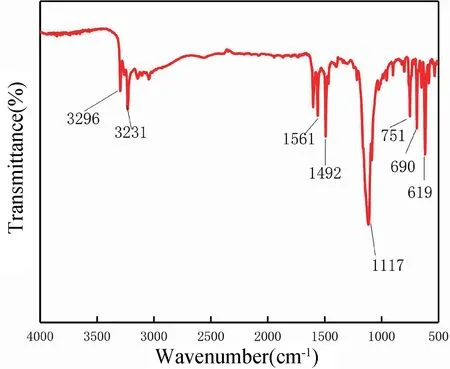
圖5 PANI 層的FT-IR 譜圖Fig.5 FT-IR spectra of PANI layer
2.5 聚苯胺層紫外光譜表征
將PANI/Cu 集流體與Bare/Cu 集流體做了固體紫外吸收光譜對比實驗,測試結果如圖6 所示。
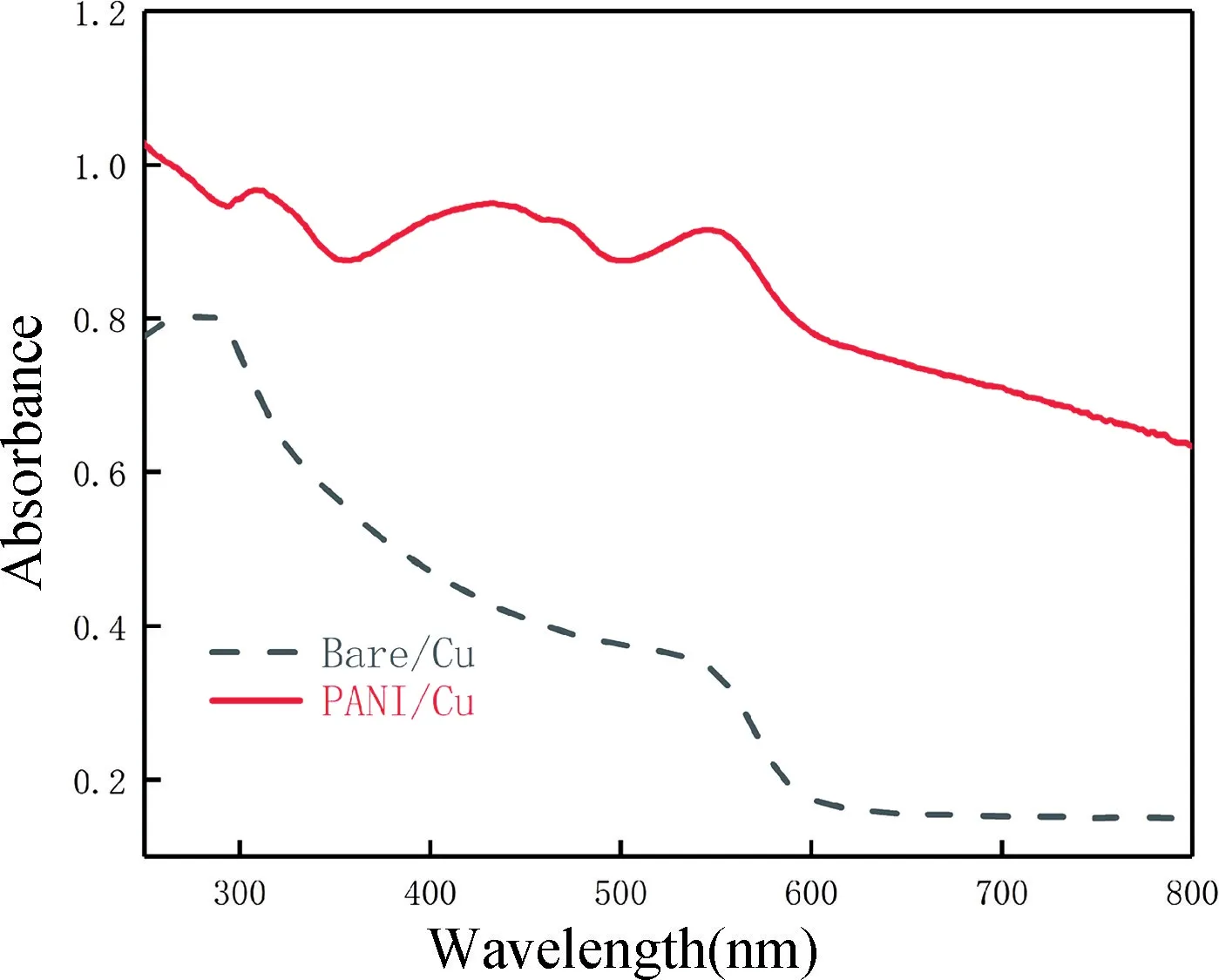
圖6 PANI 層的紫外光譜圖Fig.6 UV spectra of PANI layer
由圖6 可知,在紫外-可見光譜中觀察到了特征吸收波長區域分別在300~400,400~500,500~600 nm區域內,表明導電態形式的聚苯胺顆粒層存在。苯式結構的π-π*躍遷一般在300~400 nm 波長范圍。極化子π-π*躍遷在400~500 nm 的波長范圍,意味著中間態(氧化還原態)聚苯胺的形成是極化子的躍遷。而在500~600 nm 是第三個波長范圍極化子的π-躍遷[27-31]。綜上所述,在銅箔表面通過恒定電壓電化學聚合得到的聚苯胺顆粒層可導電,可以實現電子轉移。
2.6 電化學性能分析
2.6.1 首次充放電平臺
圖7 為聚苯胺復合銅箔集流體改性前后構成鈦酸鋰電池首次充放電結果。通過比較PANI/Cu 電池與Bare/Cu 電池在1C 倍率下的首次充放電平臺曲線可知,PANI/Cu 電池充放電比容量更高,PANI/Cu 電池首次放電比容量是185 mAh/g(LTO 的理論比容量為175 mAh/g)。這是由于PANI/Cu 首次放電時存在第二放電平臺(約1.16 V),PANI 參與嵌鋰反應并提供額外的容量,使得PANI/Cu 電池的首次放電比容量超過理論比容量。在圖7 中,Bare/Cu 電池的電壓滯后小于PANI/Cu 電池的電壓滯后,Bare/Cu 電池的放電平臺與充電平臺之間的電壓差小于PANI/Cu 電池的,原因是在LTO 上PANI 層的電化學反應出現在高電壓和低電壓[32],使得PANI/Cu 的電壓滯后增大。
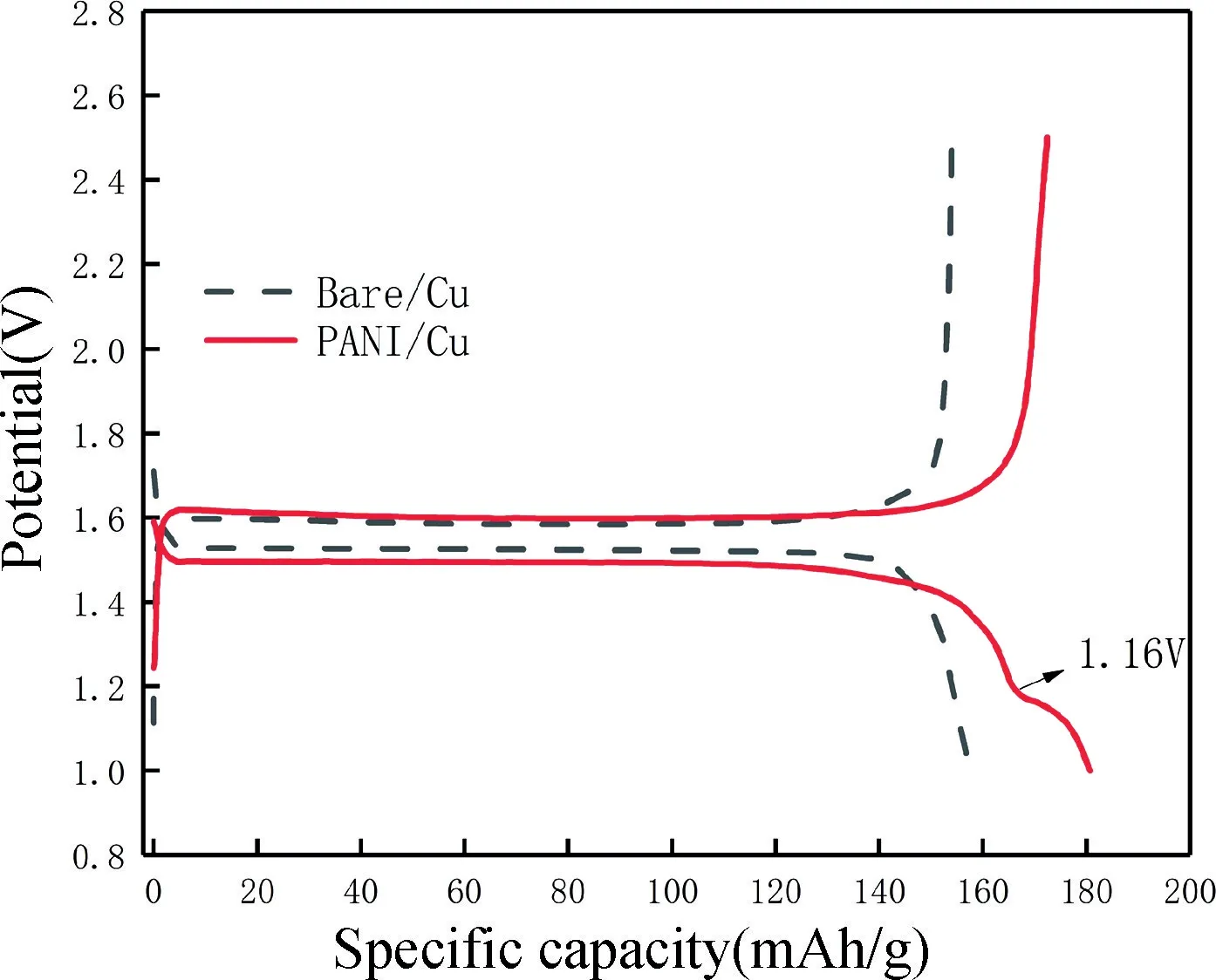
圖7 電池首次充放電平臺Fig.7 Charging and discharging platform for the first cycle of battery
2.6.2 電池在不同充放電循環次數下的充放電平臺分析
聚苯胺復合銅箔集流體改性前后構成鈦酸鋰電池在1C 倍率充放電不同循環次數下的平臺結果見圖8。圖8 結果顯示,PANI/Cu 電池在首次放電時有兩個放電平臺,該平臺的產生主要是由于PANI 層對鋰離子的電化學活性而產生的。
從圖8 可看出,PANI/Cu 電池在首次充放電平臺中出現的第二放電平臺消失,第2 圈放電比容量為174.1 mAh/g,在后續的循環過程中沒有出現超理論容量現象,表明嵌鋰初始反應過程中一部分鋰結合緊密無法脫嵌,但其后續嵌入/脫嵌反應可逆性優異。對比圖8 中(a)與(b)的充放電平臺可知,在第1,2,3,100,300,500 圈中,PANI/Cu 電池的充放電平臺相對于Bare/Cu 電池變化更具有規律性。
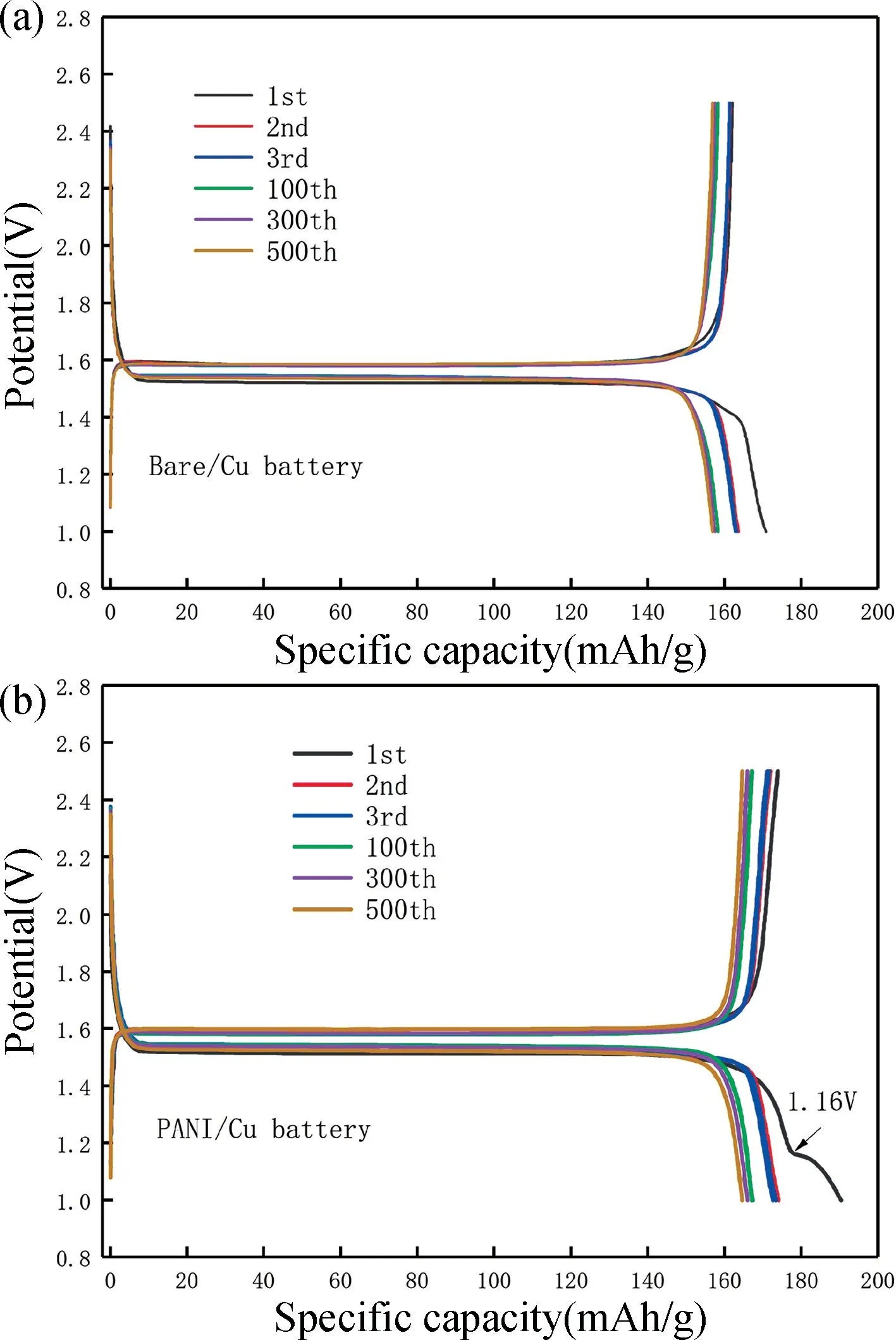
圖8 1C 倍率下不同循環數的充放電曲線。(a)Bare/Cu 電池;(b) PANI/Cu 電池Fig.8 Charge-discharge curves at different cycles under 1C rate.(a)Bare/Cu battery;(b) PANI/Cu battery
在1~500 圈循環中,PANI/Cu 電池的充放電曲線變化比Bare/Cu 電池更小,這表明PANI/Cu 電池在循環充放電過程中電極可逆性、穩定性都較好。最后,對比圖8(a)與(b)可看出,在相同的循環圈數下,PANI/Cu 電池的電壓滯后皆比Bare/Cu 電池大,但并不影響PANI/Cu 電池比Bare/Cu 電池的充放電平臺更穩定。
2.6.3 CV 首次掃描
聚苯胺復合銅箔集流體改性前后構成電池在0.5 mV/s 掃速下的首次CV 結果如圖9 所示。
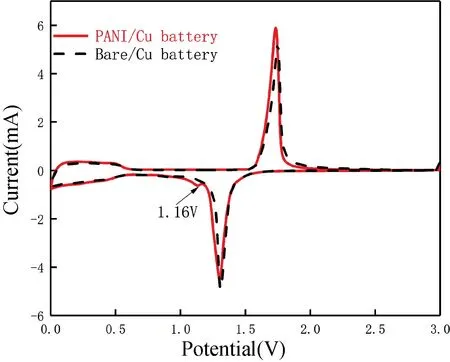
圖9 首次循環伏安性能Fig.9 First cyclic voltammetry performance
由圖9 可知,在1.7 V 與1.4 V 附近能觀察到Bare/Cu 電池與PANI/Cu 電池的氧化還原峰峰形相似,但PANI/Cu 電池在首次還原掃描時,于1.16 V處多了一個還原峰,驗證了圖7 中首次充放電平臺中的第二放電平臺。對比氧化峰與還原峰之間的電壓差,可以看出PANI/Cu 電池峰值差更大,驗證了圖7 中出現的電壓滯后現象。
2.6.4 不同CV 掃描圈數結果分析
在聚苯胺復合銅箔集流體改性前后構成電池在1C倍率情況下,不同圈數的CV 循環掃描結果見圖10。由圖10(a)與(b)可知,隨著第1 圈到第5 圈掃描次數的增加,PANI/Cu 電池的CV 曲線重合度比Bare/Cu電池更高,這表明PANI/Cu 電池的電極反應可逆性比Bare/Cu 電池的更好。再次,PANI/Cu 電池還原掃描中1.16 V 附近的還原峰在第2 圈循環時消失,正好與圖7(b)的放電平臺的消失相對應。
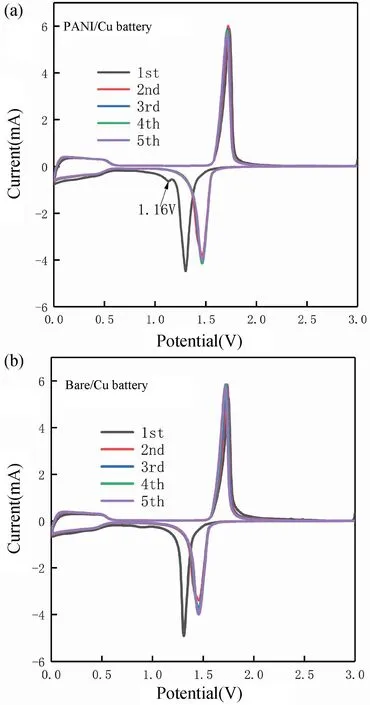
圖10 不同圈數的循環伏安性能。(a)PANI/Cu 電池;(b)Bare/Cu 電池Fig.10 Cyclic voltammetry performance at different cycles.(a) PANI/Cu battery;(b) Bare/Cu battery
2.6.5 循環性能
在聚苯胺復合銅箔集流體改性前后構成電池在1C與10C 倍率情況下的循環性能對比見圖11。
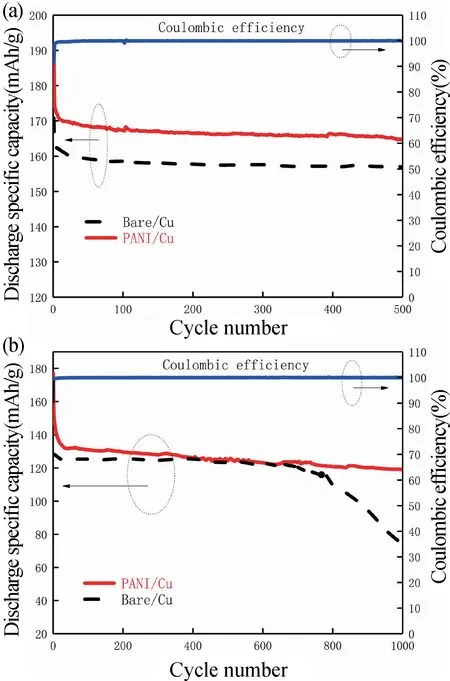
圖11 循環性能對比。(a) 1C 倍率下循環500 圈;(b) 10C 倍率下循環1000 圈Fig.11 Cycle performance comparison.(a) 500 cycles at 1C;(b) 1000 cycles at 10C
圖11(a)為PANI/Cu 電池與Bare/Cu 電池1C 倍率下分別循環500 圈的循環性能對比,圖11(b)為10C 倍率下循環1000 圈后的循環性能比對。由于鋰在反應過程中沒有不可逆的鋰消耗,因此,庫倫效率在整個過程中保持100%。數據結果表明,PANI/Cu 可以使得電池循環穩定性增強。由圖11(a)知,PANI/Cu 電池的放電比容量在1C 倍率下循環500 圈后為164.6 mAh·g-1,在相同的循環條件下,Bare/Cu 電池1C 倍率下循環500 圈后的放電比容量降到了155.6 mAh·g-1。數據結果表明PANI/Cu 電池循環穩定性比Bare/Cu 更好。在重復充放電過程中,鈦酸鋰產生無序化和結構松弛,以及越來越多Ti3+和Li+脫嵌在LTO中破壞材料結構,造成離子和電子電導率下降和極化擴大,導致容量衰減。因此,在1C 倍率500 圈循環后,兩種電池容量都發生了衰減,但是PANI/Cu 電池的放電比容量比Bare/Cu 電池的高。
如圖11(b)為在大倍率10C 下循環1000 圈測試兩種電池的循環性能。顯然,1000 圈循環后PANI/Cu 電池的容量保持率是80%,遠高于Bare/Cu 電池的60%。因此,集流體表面PANI 層的存在有效提高了LTO 電池的循環穩定性。
2.6.6 倍率性能
聚苯胺復合銅箔集流體改性前后構成電池在0.5C~10C 之間倍率性能對比見圖12。由圖12 可知,在相同倍率下,PANI/Cu 電池體系的放電比容量皆高于Bare/Cu 電池體系的放電比容量。此外,在大倍率10C 循環后突然降到0.5C 放電,Bare/Cu 和PANI/Cu電池都能瞬間恢復到最開始的放電比容量。這說明了容量更高的PANI/Cu 電池與Bare/Cu 電池具有同樣的循環穩定性與可逆性。
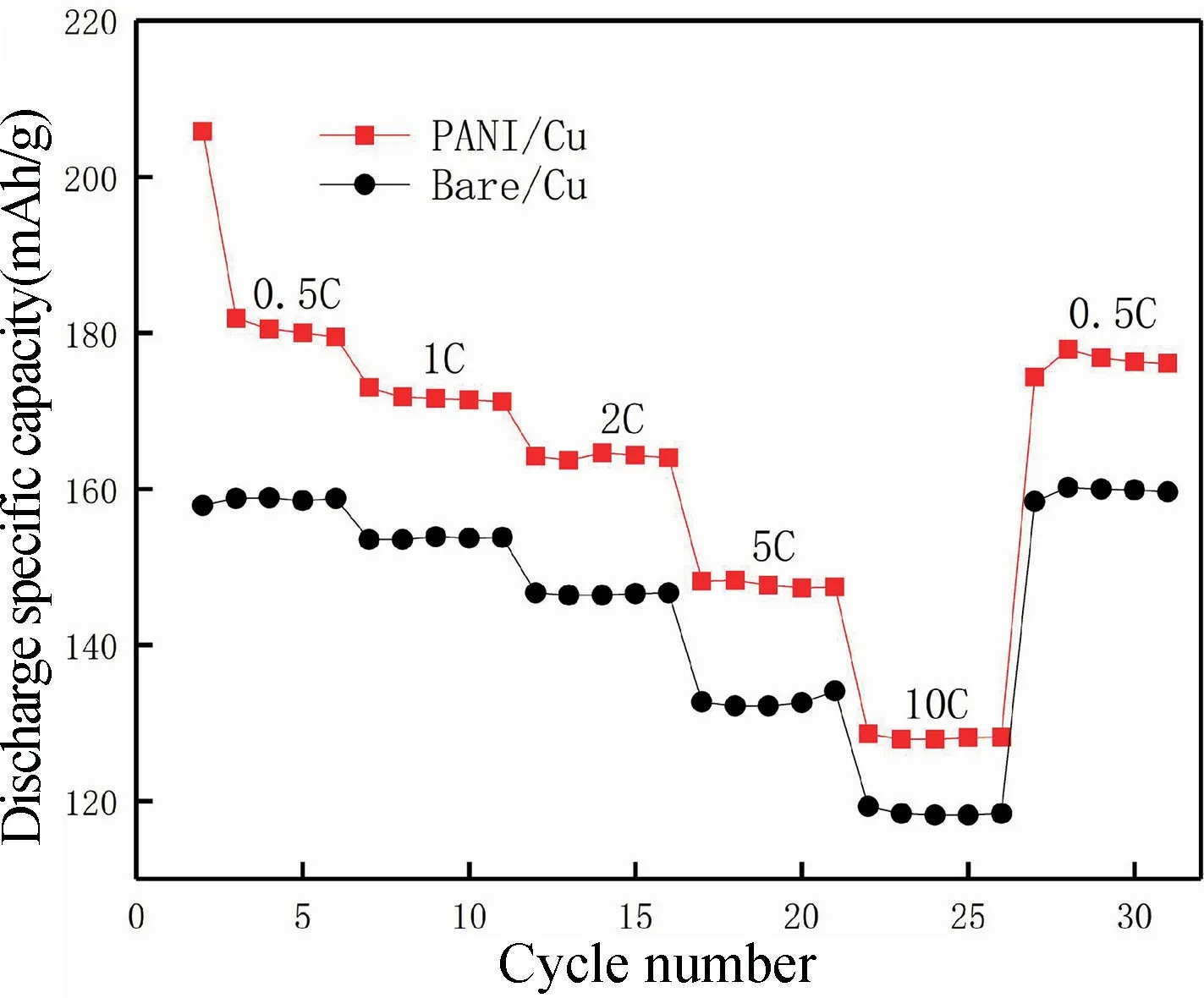
圖12 倍率放電性能Fig.12 Rate discharge performance
2.6.7 交流阻抗(EIS)
在PANI 改性前后,用LTO 電池做負極,鋰片做正極裝成電池后,交流阻抗測試對比見圖13。
圖13 為聚苯胺復合銅箔集流體改性前后構成電池的交流阻抗圖。結果表明,與Bare/Cu 組別相比,PANI/Cu 組別的電荷轉移阻抗更小。鋰離子電池阻抗的降低,是因為聚苯胺納米顆粒具有良好的導電性,提高了銅箔集流體與鈦酸鋰電池材料的兼容性,從而有效降低了電極界面阻抗,達到提高鋰離子電池的電化學性能的目的。
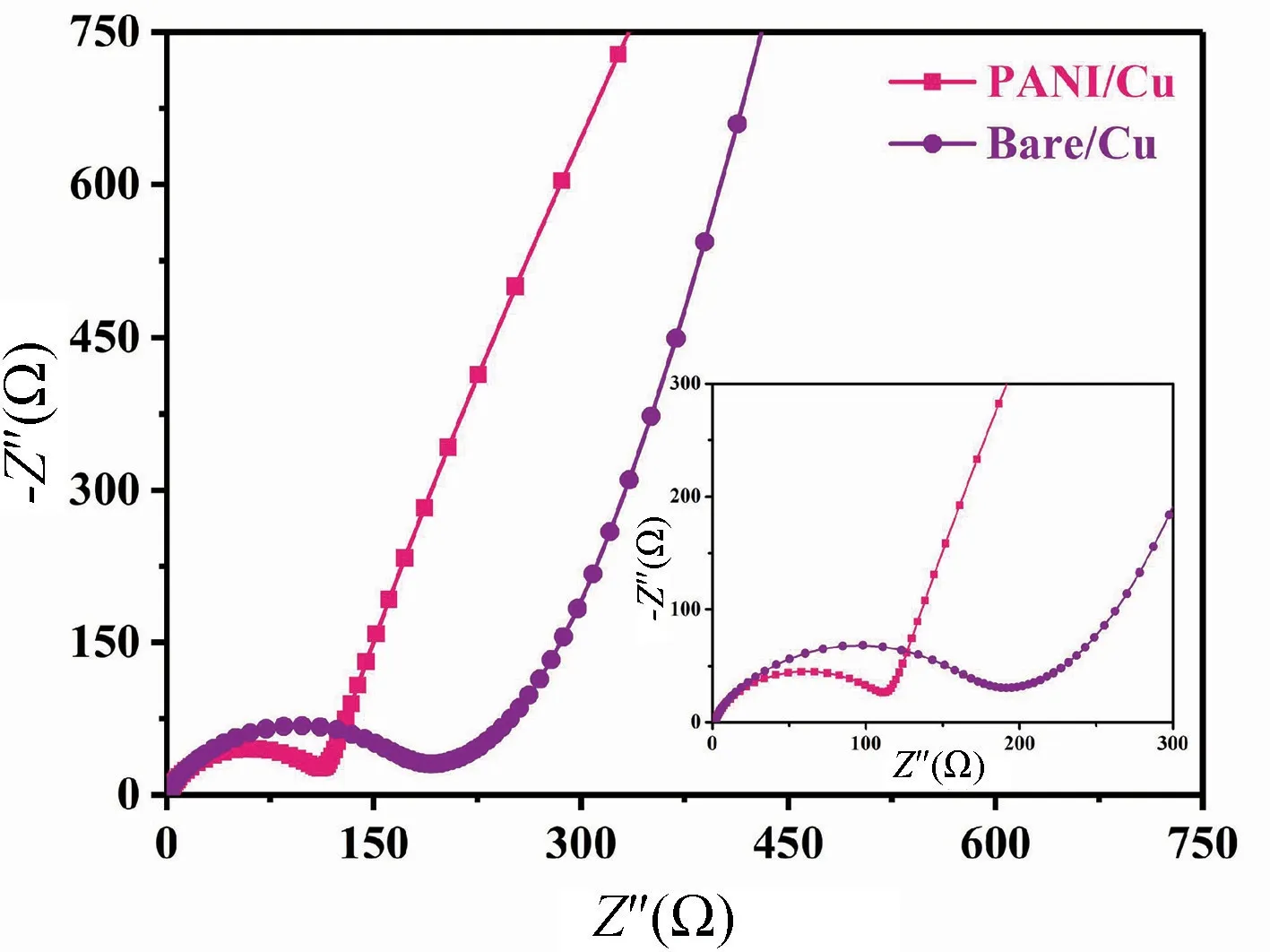
圖13 交流阻抗測試Fig.13 AC impedance measurement
3 結論
綜上所述,通過恒壓法在銅箔集流體上電化學聚合制備得到了聚苯胺薄膜,組裝成LTO 電池后通過各種性能測試,形成以下結論:
(1)在聚合的過程中,電壓對聚苯胺的形貌特征產生重要的影響,當工作電壓為1.4 V,電解質為0.05 mol/L H2SO4和0.2 mol/L 苯胺構成的混合電解質條件下,聚合10 min 后能得到均勻致密的聚苯胺復合層;
(2)FT-IR、UV-Vis 測試皆表明PANI 以導電形式存在;
(3)集流體復合改性之后,鈦酸鋰電池的循環性能及容量有所提高。在1C 倍率下充放電平臺顯示,PANI/Cu 電池首次放電比容量為185 mAh·g-1,高于純LTO 電池的理論比容量(175 mAh·g-1),而Bare/Cu 電池的放電比容量只有160 mAh·g-1左右,說明聚苯胺能夠起到嵌鋰的功能,提高了電池容量。
(4)聚苯胺對銅箔集流體的改性提高了鈦酸鋰電池在高倍率情況下的循環性能。在1C 倍率下充放電500 圈以后,PANI/Cu 電池的放電比容量為164.6 mAh·g-1,Bare/Cu 電池在相同的循環條件下,比容量卻降到了155.6 mAh·g-1,在10C 大倍率下充放電1000 圈以后,PANI/Cu 電池的容量保持率為80%,超過Bare/Cu 電池的60%,銅箔集流體表面聚苯胺改性提高了電池循環性能。
