7例Gilbert綜合征臨床分析及文獻復習
2022-10-28 07:37:38古再麗努爾吾甫爾李雪國毛敏
世界最新醫學信息文摘 2022年45期
古再麗努爾·吾甫爾,李雪國,毛敏
(新疆維吾爾自治區人民醫院血液病科,新疆 烏魯木齊830000)
0 引言
Gilbert綜合征(GS)是一種最常見的膽紅素葡萄糖醛酸化遺傳性疾病。臨床癥狀輕微,可表現為輕度、非結合型高膽紅素血癥、波動性黃疸,肝臟一般無器質性改變。非結合型高膽紅素血癥的最常見病因包括膽紅素生成過多、GS以及新生兒黃疸。GS臨床需要與溶血性貧血等血液病鑒別,臨床容易誤診或漏診,多數預后良好,不需特殊治療[1]。因此我們對2018年2月至2021年10月期間我院收治確診的7例GS臨床資料進行回顧性分析,現報道如下。
1 臨床資料
1.1 一般資料
2018年1月至2021年3月在我科住院確診的7例Gilbert綜合征;男性5例,女性2例,最小年齡22歲,最大年齡49歲;漢族3例,維吾爾族2例,回族2例,病史最長30年,最短1年。見表1。
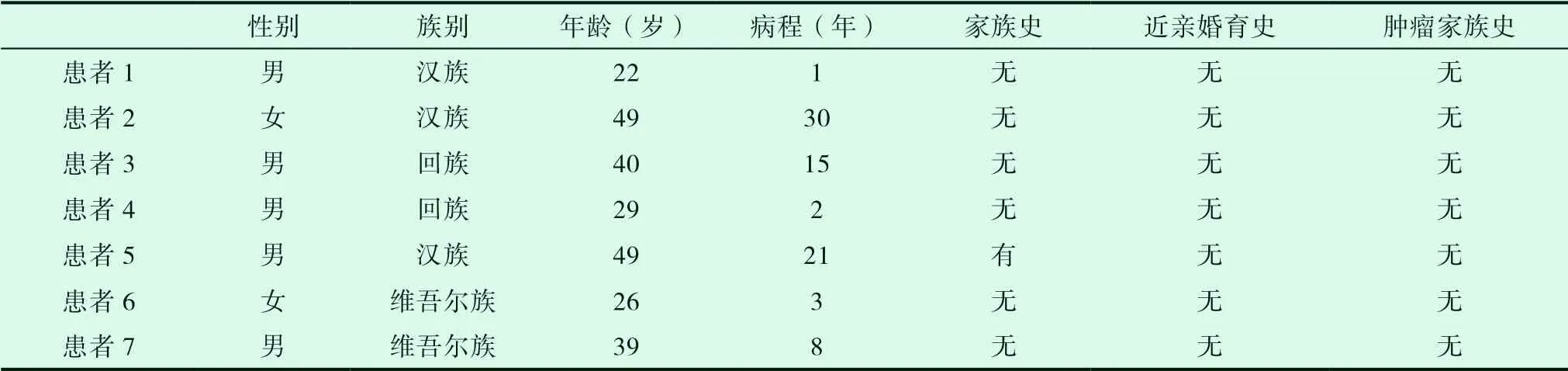
表1 患者一般情況
1.2 觀察指標
患者一般情況觀察指標一般臨床資料、血常規、網織紅細胞計數、肝功能、病毒學指標、自身免疫抗體、肝炎系列、腫瘤標記物、補體水平、免疫球蛋白、腹部B超,并均行骨髓檢查。
2 結果
2.1 臨床表現
7例患者均有皮膚鞏膜黃染,乏力3例,食欲下降1例,惡心1例,上腹部飽脹不適1例。皮膚黏膜黃染誘發因素有:勞累4例,受涼2例,感染1例,飲酒1例。
2.2 輔助檢查結果
7例均有總膽紅素、間接膽紅素輕、中度升高,其余肝功能指標均正常。甲、乙、丙肝病毒及CMV、EBV等病毒檢查均陰性;ANA、ENA、dsDNA、SMA、AMA等抗體陰性; 2例患者網織紅細胞計數增高,余檢測指標均未見異常。7例骨髓涂片和骨髓病理未見異常。腹部B超檢查7例患者肝臟、膽囊未見明顯異常。見表2。
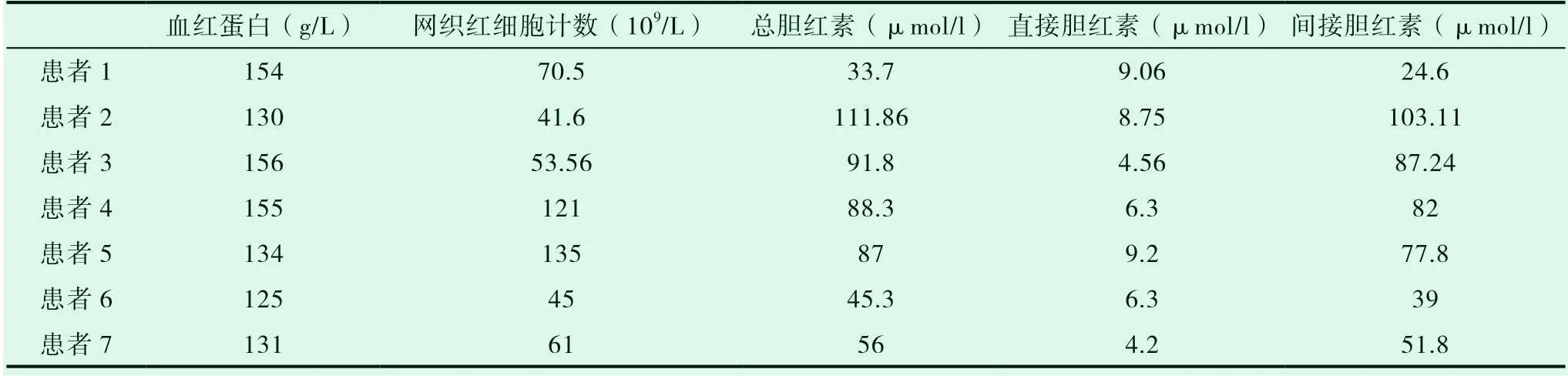
表2 血紅蛋白、網織紅細胞計數、膽紅素水平
2.3 UGT1A1突變基因位點
7例患者均進行UGT1A1基因檢測,本檢測用PCR和基因測序的方法檢測標本中UGT1A1的基因編碼區1-5外顯子以及基因上游苯巴比妥反應增強元件(PBREM)的突變分析,涵蓋了該范圍內的點突變、插入和缺失型突變。共進行5個PCR擴增反應和19個基因序列測定反應,7例患者均發生UGT1A1突變基因,見表3。
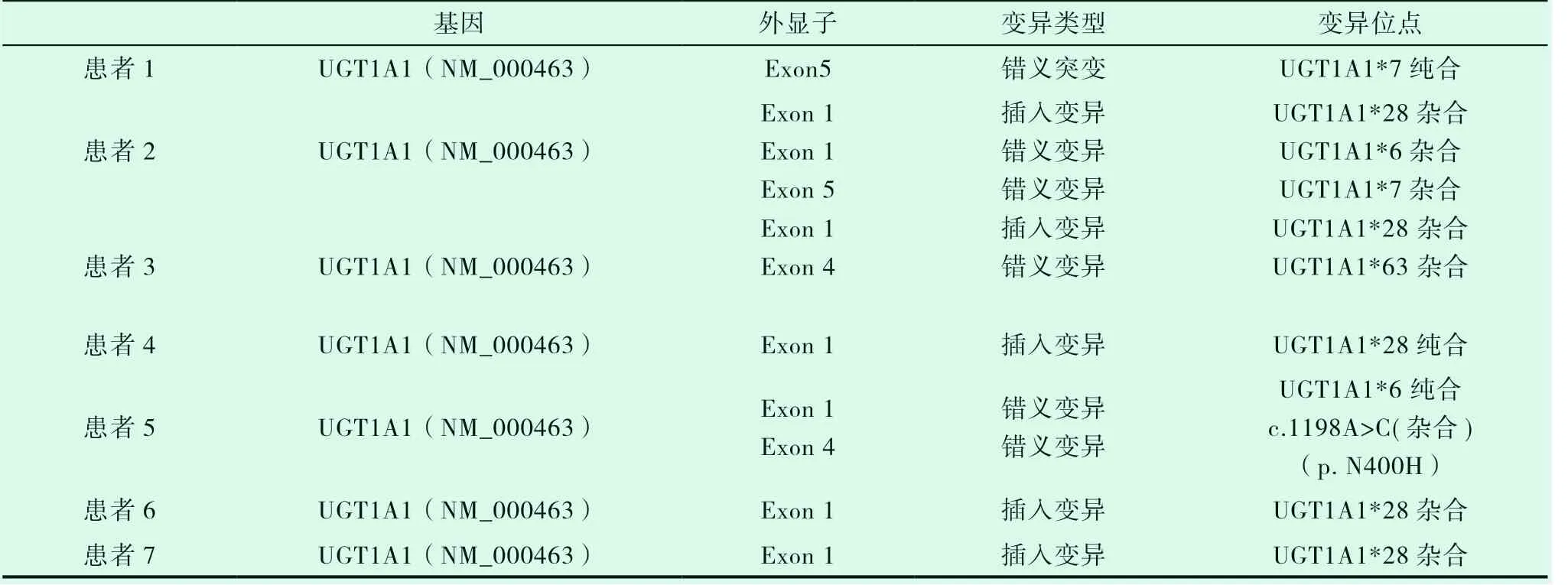
表3 UGT1A1突變基因位點檢測結果
7例患者中4例為UGT1A1*28雜合突變,該位點為突變熱點。患者4黃疸勞累、感冒等使黃疸加重,紅細胞計數和血紅蛋白正常,但紅細胞形態小細胞低色素性,網織紅細胞計數比例增高,骨髓涂片和活檢提示紅系增生明顯活躍,溶血相關檢查結果示FHb升高,Hp輕度減低,基因檢查發現UGT1A1*28純合突變,β地中海貧血基因CD14-15雜合突變,追問家族史發現父親也有常年間歇性輕度鞏膜黃染病史,父母脾臟不大,血常規、膽紅素均正常,基因篩查發現患者父母均有UGT1A1*28純合突變,父親同時伴有β地中海貧血基因CD14-15雜合突變,但父母均未發病。患者5同時存在地中海貧血基因CD14-15雜合突變和UGT1A1基因突變,可確診為β地中海貧血(輕型)和Gilbert 綜合征同時存在。患者家系UGT1A1基因檢查結果,見圖1。
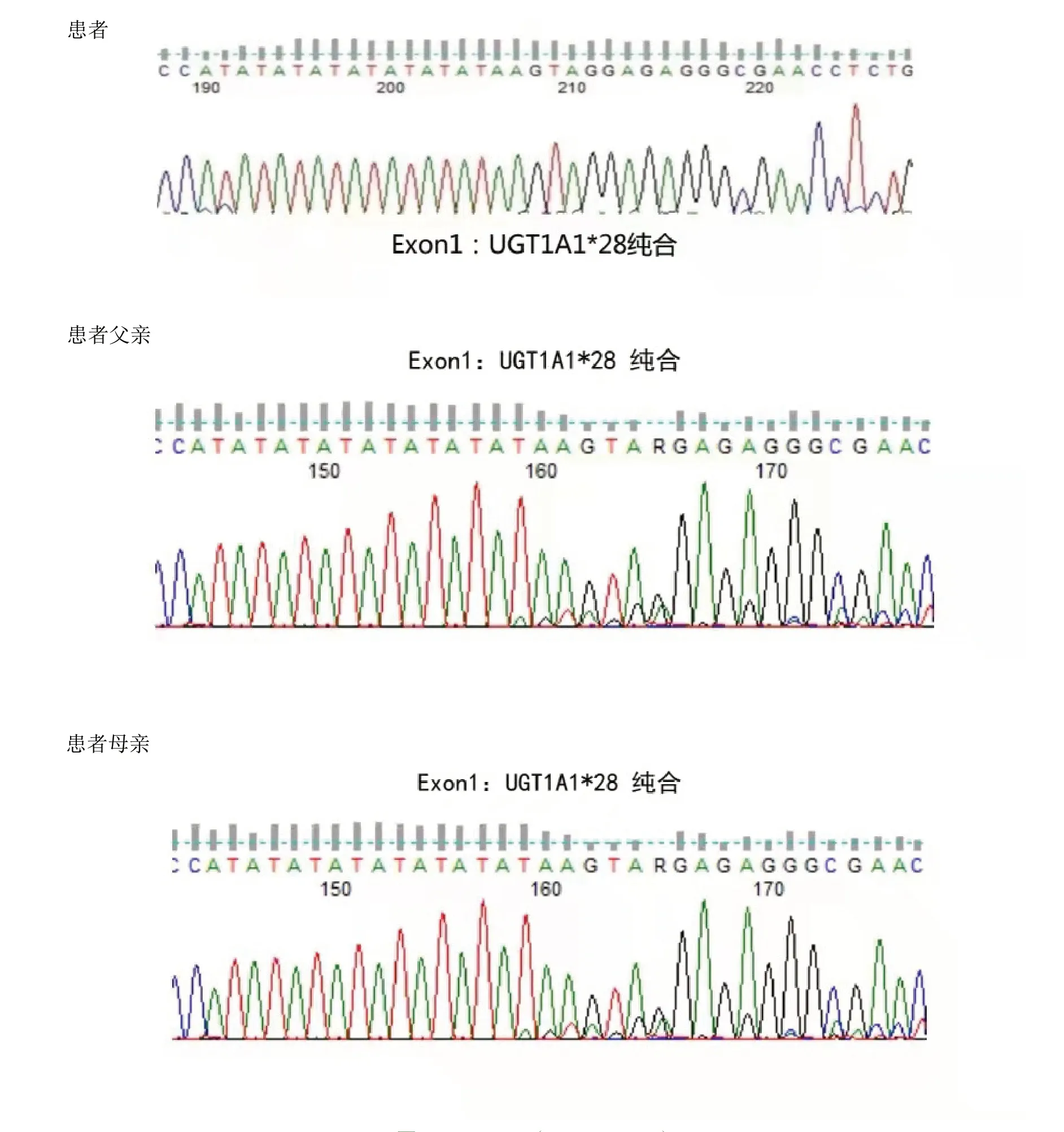
圖1 UGT1A1(NM_000463)
3 討論
Gilbert綜合征由Gilbert于1901年第1次報道,是一種膽紅素代謝障礙的常染色體隱性遺傳性疾病。GS多在青春期前后或成年期被診斷。男性多見,多有家族史。UGT1A1基因突變導致尿苷二磷酸葡萄糖醛酸轉移酶(UGT)蛋白表達水平下降,從而引起血清非結合膽紅素水平升高,出現相應臨床表現。純合子往往發病較重,而雜合子病情較輕[2]。該文中患者4為UGT1A1*28純合,患者5為UGT1A1*6純合子突變,膽紅素水平較其他患者明顯增高,與相關文獻相符。
GS的遺傳機制與位于染色體2q37位點UGT1A1基因遺傳多態性密切相關[3]。其中UGT1A1*28純和突變多見。本文中7例患者中4例存在UGT1A1*28雜合突變,一例UGT1A1*28純合突變,其余兩例分別為UGT1A1*7純合和UGT1A1*6純合突變,UGT1A1*28雜合突變為熱點突變。
盡管GS是一個良性過程,但Gilbert基因型被證實與膽石癥風險顯著相關[4],而在表型正常的人群則沒有這一風險; 另外 GS 和遺傳性球形紅細胞增多的復合遺傳也增加了膽結石的風險。患者4黃疸勞累、感冒等使黃疸加重,輕度小細胞低色素性貧血貧血,網織紅細胞計數比例增高,黃疸以間接膽紅素升高為主,骨髓涂片和活檢提示紅系增生明顯活躍,檢查結果示FHb升高,Hp輕度減低,基因檢查發現UGT1A1*28錯義突變,β地中海貧血基因CD14-15雜合突變,父親也有間歇性鞏膜黃染病史,父母脾臟不大,血常規、膽紅素均正常,父母均有UGT1A1*28純合突變,父親也發現同時伴有地中海貧血基因CD14-15雜合突變,但均未發病。該患者同時存在地中海貧血基因CD14-15雜合突變和UGT1A1基因突變,可確診為β地中海貧血(輕型)和Gilbert 綜合征同時存在。國內李露鋒[5]等報道過一例中間型ɑ-地中海貧血同時攜帶UGT1A1*28突變病例及其家系報告案例,先癥者患者的父親和女兒均缺失一個ɑ地貧基因,均為ɑ地貧,其父親網織紅細胞略高,女兒為UGT1A1*28攜帶者,但二者膽紅素均正常,其母親缺失了2個ɑ地貧基因,為輕型地貧。Lee HJ[6]等報告一例28歲男性患有膽結石和脾腫大,合并遺傳性球形紅細胞增多癥(HS)和GS的患者。國內Jun Jiang[7]等報道過一例同時存在Dubin-Johnson syndrome和GS的突變的雙重遺傳性黃疸患者,國內僅報道1例。本文中患者4目前隨訪兩年,血紅蛋白正常,無合并癥,一般情況,仍在隨訪當中。
Gilbert綜合征患者一般不需特殊治療,及時的診斷也有助于避免不必要的過度治療。因UGT1A1酶活性降低,Gilbert綜合征患者對某些藥物的敏感性增強,易產生藥物性肝損傷。國外研究認為UGT1A1*28能改善霍奇金淋巴瘤的預后、降低子宮內膜癌的風險[8]。本文中2例膽紅素明顯增高患者予以根據膽紅素水平定期口服熊去氧膽酸膠囊降膽紅素治療后膽紅素下降,余患者未予以特殊治療,病情穩定。
綜上,Gilbert綜合征是臨床上最為常見的一種先天性黃疸,對于膽紅素增高、但無溶血患者需排查GS,UGT1A1基因測序檢測是特異性及敏感度高的有效手段,可降低臨床黃疸誤診誤治,節約醫學資源。對于高膽紅素血癥患者,全面篩查相關潛在因素非常必要,在臨床表現無法用一元論解釋的時候,也應該從多元論角度啟動相關全面檢查。
