軟模板劑添加時間對SAPO-34分子篩的合成及MTO催化性能的影響
2022-11-21 13:57:12楊晨芳
天津工業大學學報 2022年5期
關鍵詞:產品
李 淵,楊晨芳
(1.天津工業大學 省部共建分離膜與膜過程國家重點實驗室,天津300387;2.天津工業大學化學工程與技術學院,天津300387)
多級孔分子篩催化劑作為一種優良的固體酸催化劑,在煉油和石化工業中得到了廣泛的應用,近年來引起了科研人員的廣泛關注。多級孔分子篩不僅具有傳統微孔分子篩優良的水熱穩定性、擇形性和可控的酸度,而且具有介孔材料的較高比表面積和傳質性能[1]。SAPO-34因其具有孔徑小、酸度適中等特點,被認為是甲醇制烯烴(MTO)工藝中最適合的催化劑。然而,在MTO進程中,微孔結構表現出嚴重的擴散限制,從而產生積炭,導致催化劑失活。多級孔SAPO-34催化劑的制備有望解決這一問題。
到目前為止,研究人員已經通過各種方法合成出多級孔SAPO-34分子篩催化劑,如硬模板法、軟模板法、無模板劑法和后合成方法等。其中,使用軟模板劑法合成催化劑不僅可以避免后合成法對分子篩骨架結構造成的破壞,而且比硬模板法操作更簡便,實驗過程也更加簡化。此外,軟模板劑通常具有較高的柔韌性和與分子篩前驅體更好的相容性,因此,軟模板法合成分子篩得到了廣泛的研究。一般采用傳統表面活性劑、兩親性有機硅烷和高聚物等作為軟模板劑[2-3]。Chen等[4]使用四乙基氫氧化銨(TEAOH)作為微孔結構導向劑,并用工業季銨型有機硅烷表面活性劑十八烷基二甲基氯化銨(TPOAC)作為介孔結構導向劑,成功合成具有特殊納米片組裝形態的多級孔SAPO-34。Bakhtiar等[5]在合成過程中添加十六烷基三甲基溴化銨(CTAB)作為晶體生長劑,通過水熱合成路線合成亞微米尺寸的SAPO-34晶體,實驗結果表明,隨著CTAB添加量的增加,晶體尺寸呈V形變化,并且當初始凝膠中CTAB與Al2O3摩爾比為0.02時,所合成的樣品性能最佳。Wang等[6]用質子化的聚乙烯亞胺(PEI)合成出分級SAPO-34,并發現介孔體積和介孔孔徑可通過PEI的分子質量和用量來調節;在酸性條件下,PEI鏈通過氫鍵和范德華力等非共價鍵與溶膠-凝膠體系中的氧化物相互作用,并在煅燒后沿PEI主鏈會形成相互連接的連續中孔;相反,在堿性條件下,PEI的卷曲減小了PEI與沸石前驅體分子的相互作用,不利于PEI在沸石基體中的包封作用。Razavian等[7]采用自組裝策略,以聚乙二醇(PEG)為介孔結構導向劑,合成具有分層網狀結構的SAPO-34分子篩,發現PEG不僅可以作為晶體生長抑制劑來調控晶體的大小,還可以作為介孔結構導向劑誘導介孔進入多孔結構。
聚乙二醇毒性低,是一種高柔性的水溶性線型聚合物,被認為在分子篩的成核和晶體生長中起著至關重要的作用。以聚乙二醇為結晶抑制劑和介孔導向劑合成多級孔SAPO-34分子篩引起了研究者的關注,但所發表的文獻中不曾提到PEG的不同添加時間對所合成樣品的影響。PEG作為結晶抑制劑和介孔導向劑,在分子篩晶體晶化的過程中對晶體的生長或溶解會產生不同程度以及不同效果的影響。結合SAPO-34分子篩的生長特性,本文通過變化其添加時間,制備出更高性能的催化劑,并探究不同軟模板劑添加時間對樣品性能的影響。
1 實驗部分
1.1 實驗藥品與儀器
藥品:擬薄水鋁石,淄博百大化工有限公司產品;磷酸,天津市化學試劑三廠產品;硅溶膠,青島海洋化工有限公司產品;四乙基氫氧化銨(TEAOH),鎮江潤景高純化工科技有限公司產品;三乙胺(TEA)、聚乙二醇(PEG1500),天津江天化工化學試劑科技有限公司產品;甲醇,天津光復科技發展有限公司產品。
儀器:FA2004型精密電子天平,上海舜宇恒平科學儀器有限公司產品;DF101S型集熱式恒溫加熱磁力攪拌器,山東菏澤正紅科教儀器有限公司產品;高壓反應釜(100 mL),天津凱易達儀器設備銷售有限公司產品;MR-A-7型微反與膜型反實驗裝置,天津大學北洋化工實驗設備公司產品;101-4AB型電熱鼓風干燥箱,天津市泰斯特儀器有限公司產品;GC-6800型氣相色譜儀,山東瑞虹化工儀器有限公司產品;MFL-2201型馬弗爐,天津市華北實驗儀器有限公司產品;DT5-2型低速臺式離心機,北京時代北利離心機有限公司產品;HJ-9型磁力加熱攪拌器,山東鄄城華魯電熱電器有限公司產品;ARL PERFORM’X型X射線熒光光譜儀,美國Thermo Fisher Scientific公司產品;Rigaku Ultima IV型X射線衍射儀,日本理學株式會社產品;ZEISS MERLIN Compact型超高分辨率場發射掃描電鏡,德國Carl Zeiss AG公司產品;Spectrum One型傅里葉變換紅外光譜儀,美國PerkinElmer公司產品;Auto chemⅡ2920型化學吸附儀,美國Micromeritics公司產品;Autosorb-iQ-C(BET)型全自動物理化學吸附儀,美國Quantachrome公司產品。
1.2 SAPO-34分子篩的合成
SAPO-34分子篩的合成采用水熱合成路線。鋁源使用擬薄水鋁石,硅源采用硅溶膠,磷酸作為磷源,模板劑采用TEAOH和TEA,軟模板劑采用PEG1500。各物質摩爾比為1(Al2O3)∶0.2(SiO2)∶0.6(P2O5)∶1(TEAOH)∶1.5(TEA)∶50(H2O)∶0.11(PEG1500)。首先將擬薄水鋁石加入到一定量去離子水中攪拌30 min,然后在劇烈攪拌下緩慢滴加磷酸,攪拌40 min得到均勻的凝膠。在凝膠中依次加入硅溶膠、TEAOH、TEA,并攪拌一定時間保證各物料混合均勻;將得到的凝膠轉移至具有聚四氟乙烯內襯的反應釜中,置于200℃的烘箱中晶化72 h,分別在晶化時間為4 h/8 h/14 h/22 h時取出反應釜,迅速加入PEG1500,并快速再次裝進高壓釜放置于烘箱中繼續晶化;晶化結束后,將所得產物進行離心、水洗,然后在100℃下烘干4 h,最后在600℃下焙燒4 h。將所得樣品按PEG添加時間由早到晚記為ST-1、ST-2、ST-3和ST-4。
1.3 SAPO-34分子篩表征
(1)XRD分析:使用Rigaku Ultima IV型X射線衍射儀進行測試,測試條件為Cu Kα靶(λ=0.154 056 nm),掃描電壓為40 kV,掃描電流為150 mA,掃描速率為5°/min,掃描范圍為5°~50°。
(2)SEM觀察:使用ZEISS MERLIN Compact型超高分辨率場發射掃描電鏡對樣品表面形貌進行觀察,將樣品均勻分散到導電膠上,加速電壓為3.0 kV。
(3)FT-IR分析:使用Spectrum One型傅里葉變換紅外光譜儀進行分析,將干燥好的樣品與KBr按照1∶100的質量比在瑪瑙研缽中研磨,混合均勻后將混合物在壓片機下以10 MPa壓力壓40 s,掃描波長的范圍為400~4 000 cm-1。
(4)酸量和酸強度測定:稱取樣品0.1 g,使用Auto chemⅡ2920型化學吸附儀進行測量,500℃下在He氣氛中預處理1 h,然后降溫到80℃,通NH3吸附達飽和,再接著用He氣進行吹掃,基線調零平穩后,按照10℃/min的升溫速率升溫至600℃,使用熱導檢測器進行脫附信號采集。
(5)XRF分析:采用ARL PERFORM’X型X射線熒光光譜儀對合成的分子篩進行分析,測角儀轉速為4 800°/min,以550°/min的速率掃描元素譜線。
(6)BET分析:在Autosorb-iQ-C(BET)型全自動物理化學吸附儀上測定樣品的比表面積、孔結構和孔徑。
1.4 MTO催化反應
采用MR-A-7型固定床反應裝置測試樣品的MTO催化性能。將合成出的SAPO-34分子篩壓片、篩分,得到20~30目的顆粒,稱取出1 g分子篩顆粒填入反應器的恒溫段(尺寸:380 nm×10 nm×1.5 mm)。預熱溫度和反應溫度分別為160℃和460℃,進樣參數為進料空速5 h-1且原料中水質量分數為60%。反應產物利用氣相色譜進行分析,將從反應開始到甲醇的轉化率低于99%的時間定義為催化劑的壽命。
2 結果與討論
2.1 XRD分析
圖1所示為不同PEG添加時間下合成的樣品的XRD譜圖。

圖1 不同PEG添加時間樣品的XRD圖Fig.1 XRD patterns of samples synthesized by different addition time of PEG
由圖1可以看出,樣品在2θ=9.5°、12.9°、16.0°、17.7°、20.6°、24.9°、30.6°、31°處均出現較強的特征衍射峰,與CHA結構完全吻合[8],并且沒有其他晶相存在。可以看出,晶化14 h時加入PEG1500得到的樣品ST-3的衍射峰強度最大,說明相較于其他樣品,該樣品具有較高的結晶度。
2.2 SEM分析
不同PEG添加時間合成樣品的SEM圖如圖2所示。

圖2 不同PEG添加時間合成樣品的SEM圖Fig.2 SEM images of samples synthesized by different addition time of PEG
由圖2可以看出:合成的分子篩均為典型的立方結構;晶化8、14 h時加入PEG1500時得到的樣品ST-2、ST-3中存在無定形顆粒,并且晶體表面存在缺陷且尺寸不均一,這可能是添加的PEG1500在體系中分布不均勻造成的;晶化4、22 h時加入PEG1500得到的樣品ST-1、ST-4為表面規整的立方結構,尺寸較為均一。此外結合圖2,利用Nano Measurer粒度分析軟件對合成樣品的粒徑進行估測,結果如圖3所示。
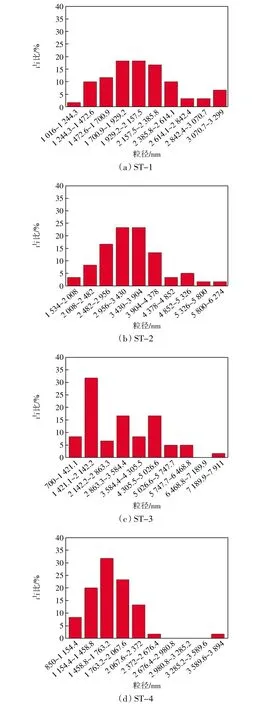
圖3 不同樣品粒徑分布Fig.3 Particle size distribution of different samples
由圖3計算,得到4個樣品中尺寸占比最大的顆粒,其尺寸分別為1 930、2 956、1 781、1 611 nm。可以看出合成的多級孔SAPO-34分子篩晶體尺寸在1~2 μm之間,只有樣品ST-2的晶體尺寸大于2 μm。
通常認為,分子篩的晶化過程是一個動態過程,期間伴隨著晶體的生長與晶體的溶解。晶化過程中加入的PEG1500會與體系中物質發生相互作用,從而對分子篩晶體的生長過程以及溶解過程產生影響。反應進行22 h后添加PEG1500,隨著反應時間的增加,體系中的活性成分逐漸減少,軟模板劑分子與體系中較少的活性成分發生了較強的相互作用,致使鋁物種和硅物種不能有效及時供應,即此階段添加的PEG1500對晶體的生長階段產生了抑制作用,因而得到了顆粒尺寸較小的分子篩樣品。晶化8 h后加入PEG1500得到的樣品ST-2粒徑較大的原因可能是由于該階段加入的PEG1500與前驅體的活性位點發生相互作用,這些前驅體在PEG1500的導向作用下生成了較少的晶核,所以得到了粒徑較大的SAPO-34樣品。PEG的添加增加了合成介質的粘度和不均勻性,抑制了鋁和硅物種的轉移。此外,乙二醇聚合物的羥基陰離子官能團與硅醇基團(通過氫鍵作用)和鋁陽離子(通過螯合作用)相互作用,限制了陽離子的轉移。由于陽離子的低轉移率和傳質的減少,鋁物種和硅物種不能及時供應,從而晶體的成核和生長受到抑制。因此,PEG導向合成的晶體核數較少,粒徑較大[9]。在合成體系中加入聚乙二醇,PEG鏈與前驅體的活性位點發生相互作用,占據了晶核表面,從而抑制了晶體的聚集,也可以說PEG鏈增大了單晶間的靜電斥力[9-10],通常這種情況下會得到粒徑較小的產物。然而,PEG分子與原料相互作用得到的合成介質類型、前驅體中形成的PEG膠束的數量和大小以及結晶條件都是影響不同PEG添加時間對SAPO-34分子篩結構調節作用的重要因素[11]。
2.3 FTIR分析
圖4為合成樣品的FTIR圖。
由圖4可以看出,本文所合成的樣品具有相似的紅外譜圖,出峰位置與Amoozegar的報道[12]符合。530 cm-1處為PO4四面體P-O的彎曲振動峰;640 cm-1處對應骨架雙六元環振動峰;730 cm-1處為Al-O(或P-O)的對稱振動峰;1 110 cm-1處對應O-P-O的非對稱振動峰;P-O-P(或P-O-Al)的非對稱振動峰在1 215 cm-1處[13-14]。此外,4個樣品在2 400~3 000 cm-1處未出現紅外吸收峰,表明樣品中不存在游離態的磷酸以及模板劑分子,則可以說明全部磷酸均參與到反應中,并且在600℃下對樣品進行焙燒可以完全除去有機模板劑。3 450 cm-1處對應與B酸位有關的橋連羥基(Si-OH-Al)的拉伸振動峰,由圖4可見,樣品ST-2在此處吸收峰強度最大,即說明ST-2具有最高的強酸量;樣品ST-1在此處的吸收峰強度較弱,說明樣品ST-1的強酸量較低。

圖4合成樣品的FTIR圖Fig.4 FTIR patterns of synthesized samples
2.4 NH3-TPD分析
圖5 為合成樣品的NH3-TPD譜圖。

圖5 合成SAPO-34的NH3-TPD譜圖Fig.5 NH3-TPD patterns of synthesized SAPO-34
由圖5可以看到,所合成的4個SAPO-34樣品均呈現2個脫附峰,這與文獻[15]報道的SAPO-34分子篩的酸性質相符。通過NH3脫附峰出現的溫度高低可以分析SAPO-34分子篩表面酸性位點的強弱,可以觀察到,4個產品的譜圖相差不大,低溫、高溫解吸分別在100~300℃、300~500℃。100~300℃對應分子篩的弱酸位點,弱酸位的產生是P-OH的羥基基團與四面體AlO4相互作用而產生的;300~500℃對應分子篩的強酸位點,強酸位的產生是因為Si進入分子篩骨架[16-18]。幾種催化劑弱酸酸位的強度和酸量相差不大,強酸量和強酸強度存在略微明顯的差異。其強酸量順序為:ST-2>ST-3>ST-1>ST-4。通常認為,甲醇以及各類產物是在酸性位上發生一系列的反應[19-20],值得強調的是,酸位過多會促進烯烴的氫轉移反應,強酸位則會促進芳族反應,加速催化劑的失活。
2.5 XRF分析
對不同PEG1500添加時間合成的分子篩樣品進行XRF表征,并計算樣品的硅進入程度。硅進入程度為產品的n(Si)/n(Al+P+Si)與初始凝膠中的n(Si)/n(Al+P+Si)的比值,結果如表1所示。

表1 合成樣品的化學組成Tab.1 Chemical composition of synthesized samples
由表1可見,不同添加時間下合成的產品的元素組成大致相同。硅鋁比均在0.29左右,只有在14 h時添加PEG1500得到的樣品ST-3的硅鋁比數值約為0.33,鋁含量明顯低于其余3個樣品,其表現為結構缺陷,從圖2 SEM結果可以清晰看到該樣品呈現出不規則的立方體結構。4 h添加PEG1500所得樣品的Si含量最低,代表其B酸中心的強度或數目低于其他3個樣品[21],這與FTIR的表征結果一致。樣品ST-3的硅鋁比最大,結合NH3-TPD的測試結果,其酸強度卻弱于樣品ST-2的酸強度,猜測這種現象產生的原因可能是樣品ST-3中存在大量的“硅島”,所以其酸強度減弱。
2.6 BET分析
圖6為標準溫度及壓力狀態下樣品ST-1和ST-4的N2吸脫附曲線。
由圖6可以看出,樣品ST-1和ST-4的N2吸脫附曲線特征類似于具有典型微孔結構的傳統SAPO-34分子篩,但是與典型的Ⅰ型等溫線略有不同,其表現為Ⅰ+Ⅳ型氮氣吸附等溫線,可以明顯觀察到在0.6<P/P0<1.0的壓力區域范圍內出現了一定程度的遲滯現象,這表明樣品除微孔外,還有少量介孔孔道的存在。

圖6 ST-1與ST-4分子篩樣品的N2吸附-脫附曲線與孔徑分布Fig.6 N2 adsorption and desorption patterns and pore size distribution of molecular sieves ST-1 and ST-4
2.7 MTO催化性能評價
圖7、圖8分別為MTO反應進程的甲醇轉化率以及乙烯和丙烯的選擇性,反映合成樣品的催化性能。
由圖7可以看出,4個樣品對甲醇的最大轉化率均接近100%;樣品ST-4壽命最短,僅90 min;樣品ST-1壽命則可以達到180 min;壽命長短依次為:ST-1>ST-2>ST-3>ST-4。ST-3過低的酸量在活性位點被積碳覆蓋后難以將甲醇/二甲醚進行有效轉化,故而其壽命短,且甲醇轉化率低。ST-2具有最高的酸量,但是過高的酸量使得氫轉移反應加快,導致積碳的生成,加速催化劑的失活。由圖8可以看出,樣品ST-1表現出最高的雙烯選擇性,最高可達91.86%,樣品ST-4選擇性最差,樣品ST-2的選擇性略低于ST-3樣品的選擇性。這可能是由于樣品ST-2具有較強的酸量,促使丙烯發生氫轉移現象,從而有較低的雙烯選擇性。

圖7 SAPO-34分子篩催化MTO反應的甲醇轉化率Fig.7 Methanol conversion of MTO reaction catalyzed by SAPO-34 molecular sieve

圖8 SAPO-34分子篩催化MTO反應的乙烯和丙烯選擇性Fig.8 Selectivity of ethylene and propylene in MTO reaction catalyzed by molecular sieve SAPO-34
3 結論
利用水熱合成法,以四乙基氫氧化銨和三乙胺為混合模板劑,聚乙二醇為軟模板劑,通過改變聚乙二醇的添加時間制備出多級孔SAPO-34分子篩。結果表明:
(1)聚乙二醇作為介孔導向劑和結晶抑制劑,不僅可以在微孔結構中引入介孔,還可以起到抑制晶體結晶的作用。結合SEM結果,發現不同時間加入軟模板劑對分子篩晶體的成核和生長產生不同結果。4 h和22 h添加軟模板劑得到的產品形貌規整且不存在無定形顆粒,8 h和14 h添加軟模板劑得到的產品晶體存在缺陷且有無定形顆粒存在。NH3-TPD測試結果表明,改變軟模板劑添加時間,對分子篩的酸性會產生不同影響。
(2)通過改變軟模板劑添加時間得到一系列多級孔SAPO-34分子篩,其中4 h添加軟模板劑所合成的樣品具有較大的晶粒尺寸,適宜的酸度,在甲醇制低碳烯烴進程中表現出最長的催化壽命(180 min)以及最高的雙烯選擇性(91.86%)。
猜你喜歡
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
物流技術與應用(2022年5期)2022-06-17 06:01:38
快樂語文(2021年36期)2022-01-18 05:48:46
金橋(2021年4期)2021-05-21 08:19:22
中國化妝品(2018年6期)2018-07-09 03:12:40
中國化妝品(2018年6期)2018-07-09 03:12:32
Coco薇(2015年1期)2015-08-13 02:23:50
汽車維修與保養(2015年6期)2015-04-17 03:31:50
玩具(2009年10期)2009-11-04 02:33:14
