氨化纖維素基氣凝膠的制備及其對阿散酸的吸附行為研究
2022-11-24 05:39:12鄭雪琴吳志浩楊桂芳葉曉霞
中國造紙 2022年9期
鄭雪琴 吳志浩 楊桂芳 葉曉霞,*
(1.福建船政交通職業學院安全與環境學院,福建福州,350007;2.福州大學環境與安全工程學院,福建福州,350108;3.福建省海洋生物多樣性保護與永續利用重點實驗室,福建福州,350108;4.閩江學院海洋研究院,福建福州,350108)
芳香族有機砷化物如對氨基苯胂酸(又名阿散酸,p-ASA)具有治療球蟲腸道寄生蟲、提高飼養效率、促進畜禽快速生長和改善肉類色素沉著等作用,已被廣泛用作飼料添加劑[1-3]。畜禽攝入的p-ASA經消化后,最終隨糞便排出體外[4],通過禽畜垃圾進入環境中[5]。然而,p-ASA可通過非生物和生物轉化為有毒的無機砷[6],進入到周圍環境中造成污染,影響植物和微生物的生長發育,甚至通過食物鏈的傳遞,危及人類健康[7]。因此,為降低p-ASA的污染,需要開發一種環境友好、成本低廉的處理方式。目前,對于p-ASA的去除方法主要有氧化[8]、吸附[9]、光催化降解[10]和高級氧化降解等[11]。雖然降解法對p-ASA的降解效率很高,幾乎可達到完全降解,但是為了避免降解產物的二次污染,還需要設置后續處理過程;另外,降解過程中的催化劑制備困難、成本高、回收難度大,所以降解法在實際應用中仍存在較大的限制。在上述處理方法中,吸附法具有高效簡便的特點,同時不會產生對環境造成二次污染的中間產物,且當污染物濃度較低時,吸附劑仍可以有效地去除污染物,是目前處理p-ASA的主要方法之一[12]。近年來,錳氧化物[13]、鐵氧化物[14]、金屬有機框架物[15]等被作為p-ASA的主要吸附材料,且有一些已被證實具有良好的吸附效果,但成本高、回收難等缺點限制了這些材料在實際中的應用。為了解決這些問題,研究人員開始研究磁性復合碳材料[16]和宏觀塊狀材料[17-18]作為吸附劑的效果,特別是氣凝膠[19]和膜材料[20]等具有高吸收性和可回收性的宏觀材料引起了廣泛關注。
纖維素基氣凝膠作為獨立于無機氣凝膠和有機聚合物氣凝膠的第三代氣凝膠,具有低成本和優異性能,如密度低、比表面積大(108~1039 m2/g)、孔隙率高、廉價易回收、可生物降解和生物相容性佳等優點[21-23]。目前,纖維素基氣凝膠已被用于去除各種污染物,如金屬離子(Cu2+[24]),水產品養殖廢水中的硝酸鹽、亞硝酸鹽和磷酸鹽[25],廢水中的染料(亞甲基藍[26]、酸性紅[27]、剛果紅[28]等)。纖維素基氣凝膠不僅可以處理廢水,還可以作為捕獲劑來捕獲CO2[29]。然而,原始纖維素基氣凝膠在吸附有機污染物時僅具有羥基官能團,羥基活性遠低于其他基團(如氨基和羧基),吸附能力有限。因此,需對原始纖維素表面進行化學修飾引入活性吸附位點以增強其對有機物的吸附能力,如通過氨化、磺化、賦磁[30-32]等方式,提高纖維素基氣凝膠的吸附量、機械強度,并簡化制備工藝流程。
本研究在纖維素基氣凝膠的基礎上,以環氧氯丙烷為交聯劑,通過交聯反應和冷凍干燥制備聚乙烯亞胺(PEI)功能化纖維素基氣凝膠Cell@PEI,對Cell@PEI進行結構表征,同時通過改變pH值、吸附時間和吸附溫度等反應條件,考察Cell@PEI對p-ASA的吸附性能及吸附機理。
1 實驗
1.1 主要試劑
纖維素粉末(粒徑90 μm)、PEI(純度99%)、環氧氯丙烷(ECH)和p-ASA,均購自上海阿拉丁生物科技股份有限公司;尿素、氫氧化鈉(NaOH)、鹽酸,均購自國藥化學試劑有限公司;以上藥品均為分析純。
1.2 主要儀器
紫外可見分光光度計,T6型,北京普析通用儀器有限責任公司;Zeta電位與納米粒度儀,Nano?Plus3型,美國Micromeritics公司;傅里葉變換紅外光譜儀(FT-IR),AVAT-AR 360型,美國Nicolet公司;掃描電子顯微鏡(SEM),Quanta250型,美國FEI公司;比表面積分析儀,ASAP2020型,美國Mi?cromeritics公司;X射線衍射儀(XRD),Miniflex 600型,日本株式會社Rigaku;X射線光電子能譜儀(XPS),ESCALAB 250型,美國ThermoFisher公司;智能恒溫培養振蕩器,ZWYR-2012C型,上海智城分析儀器制造有限公司;冷凍干燥機,Lab-1A-50型,北京博醫康實驗儀器有限公司;精密增力電動攪拌器,JJ-1型,金壇市富華儀器有限公司。
1.3 吸附劑的制備
將4.0 g纖維素粉末分散在NaOH、尿素和水的混合溶液(質量比7∶12∶81)中。將混合溶液放入25℃的水浴中進行超聲處理,得到質量分數為4%的纖維素懸浮液。將懸浮液預冷卻至?13.5℃并用電動攪拌器強烈攪拌,得到透明的纖維素溶液。將50.0 g纖維素溶液和4.0 g PEI置于三頸燒瓶中混合,攪拌30 min后加入交聯劑ECH(10.0 g),將混合物在70℃條件下反應4 h,形成水凝膠,隨后經超純水置換和循環冷凍干燥(?70℃,12 h),最終制得Cell@PEI,其反應過程如圖1所示。
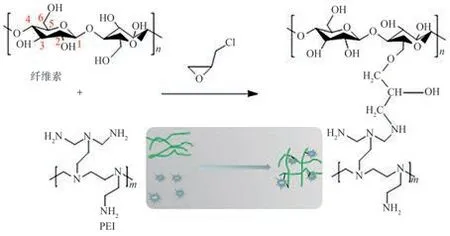
圖1 Cell@PEI的制備反應過程示意圖Fig.1 Schematic diagram of Cell@PEI preparation reaction process
1.4 分析表征
采用SEM表征樣品的表面微觀形貌;采用比表面積分析儀的BET模型表征樣品的孔徑分布和比表面積;采用XRD測定樣品晶型結構;采用FT-IR測定樣品表面官能團,對600~4000 cm?1范圍內掃描36次,擬合后得到紅外吸收光譜。
1.5 吸附實驗
吸附過程在恒溫振蕩培養箱(溫度25℃、轉速160 r/min、時間6 h)中進行,用0.1 mol/L NaOH溶液和0.1 mol/L鹽酸調節p-ASA溶液的pH值。將一定量Cell@PEI置于50 mL的p-ASA溶液(50 mg/L)中一定時間,以確保達到吸附平衡。研究吸附劑用量(0.01~0.07 g)、pH值(3~10)、p-ASA的初始濃度和共存離子對吸附參數的影響。反應后,利用紫外-可見分光光度計測定上清液中p-ASA的濃度。隨后,分別采用式(1)和式(2)計算p-ASA的去除效率(η,%)和Cell@PEI的平衡吸附量(Qe,mg/g)。
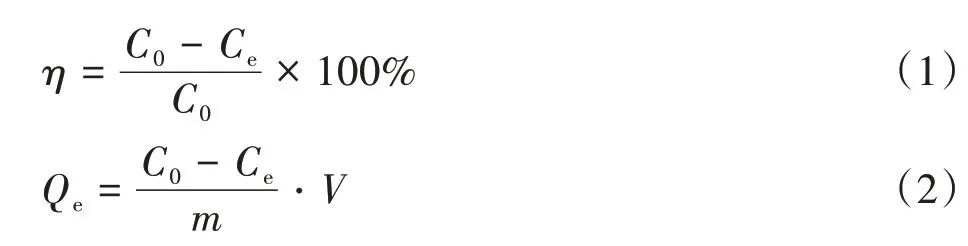
式中,C0表示溶液中吸附質的初始濃度,mg/L;Ce表示吸附質剩余濃度,mg/L;m表示吸附劑用量,g;V表示吸附質溶液體積,L。
1.6 吸附再生實驗
采用0.2 mol/L和0.5 mol/L NaOH溶液為解吸劑對吸附p-ASA后的Cell@PEI進行5次解吸再生。每次再生后抽濾、用去離子水洗滌Cell@PEI至pH值為7并干燥(70℃,12 h),重復吸附實驗同1.5部分。計算每次再生吸附后的Cell@PEI對p-ASA的去除率和Cell@PEI的平衡吸附量。
2 結果與討論
2.1 Cell@PEI的表征
2.1.1 SEM分析
利用SEM觀察纖維素粉末原料和Cell@PEI的表面微觀形貌,結果如圖2(a)和圖2(b)所示。從圖2(a)可以看出,纖維素粉末表面粗糙,呈分散狀;而纖維素與PEI交聯接枝后,制備的Cell@PEI呈疏松的多孔網狀結構,孔道結構發達(見圖2(b)),這使得Cell@PEI具有較大的比表面積,增加其與污染物的接觸面積,為p-ASA的吸附提供足夠吸附位點,從而提高吸附效率。
由吸附前后Cell@PEI的EDS圖(見圖2(c))可知,N元素存在于Cell@PEI表面上,證明氨基已經成功引入氣凝膠材料中。在Cell@PEI吸附p-ASA后,As均勻分布在其表面上,證明p-ASA被Cell@PEI成功吸附。
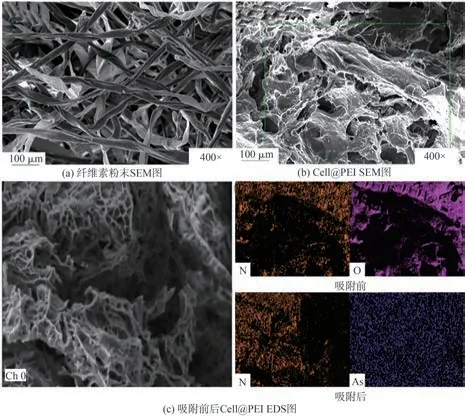
圖2 纖維素粉末及Cell@PEI的SEM圖和改性及吸附前后Cell@PEI的EDS圖Fig.2 SEM images of cellulose powder and Cell@PEI;EDS images of Cell@PEI before and after modification
2.1.2 孔結構分析
采用比表面積分析儀,根據BET模型和N2吸附-脫附方法測定Cell@PEI和纖維素粉末的孔徑分布和比表面積,結果如圖3所示。根據國際純粹與應用化學聯合會(IUPAC)公布的氣體吸附等溫線分類標準,Cell@PEI符合V型等溫線的特征,比表面積達241.4 m2/g,平均孔徑為6.27 nm,孔隙率為91%,是典型的介孔材料。與纖維素粉末相比,Cell@PEI的比表面積顯著提升,更有利于吸附污染物,這與SEM的分析結果一致。
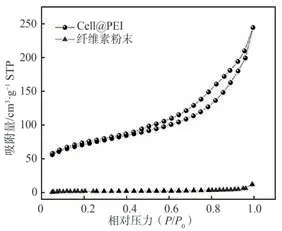
圖3 纖維素粉末和Cell@PEI的N2吸附-脫附等溫線Fig.3 N2 adsorption-desorption isotherm curves of cellulose powder and Cell@PEI
2.1.3 XRD分析
圖4為纖維素粉末和Cell@PEI的XRD譜圖。從圖4可以看出,Cell@PEI的結晶度有所下降。纖維素粉末為I型結晶結構,在2θ=15.0°、16.4°、22.6°及34.2°處出現的峰分別對應纖維素I型結構的典型晶面(101)(101)(002)及(040);而Cell@PEI的XRD譜圖呈現出典型的纖維素II型結構,其分別在2θ=20°和22°時出現II型結構的典型晶面(101)。這說明經過PEI改性后,纖維素的晶型結構發生了明顯變化,同時也說明PEI已經成功接枝到了纖維素上。
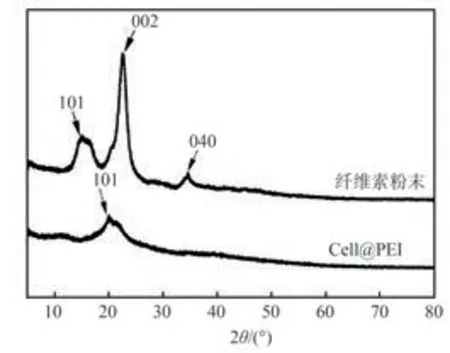
圖4 纖維素粉末和Cell@PEI的XRD譜圖Fig.4 XRD patterns of cellulose powder and Cell@PEI
2.1.4 FT-IR分析
圖5為纖維素粉末、PEI、Cell@PEI吸附p-ASA前后的FT-IR譜圖。經PEI改性的纖維素氣凝膠在2920和1030 cm?1出現的吸收峰分別與C—H和C—O鍵的拉伸振動相關,是典型的纖維素特征峰,證實PEI改性并未改變纖維素的基本官能團。此外,Cell@PEI在1601和3290 cm?1處的吸收峰與N—H拉伸振動及纖維素中的O—H拉伸振動有關。另外,從吸附后材料(Cell@PEI-p-ASA)的譜圖可以看出,與Cell@PEI相比,Cell@PEI-p-ASA在1093和826 cm?1處出現C—As和As—O的特征吸收峰,證明吸附后的Cell@PEI中存在As,p-ASA被成功吸附。
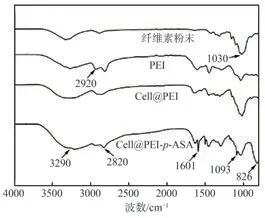
圖5 改性及吸附前后纖維素材料的FT-IR譜圖Fig.5 FT-IR spectra of cellulose materials before and after modification and adsorption
2.2 Cell@PEI對p-ASA的靜態吸附性能分析
2.2.1 pH值對Cell@PEI吸附性能的影響
圖6為不同吸附條件對Cell@PEI吸附p-ASA的影響。添加0.02 g Cell@PEI,控制p-ASA初始濃度為60 mg/L,吸附時間為250 min,改變溶液pH值,得到不同吸附量和去除率,結果如圖6(a)所示。由圖6(a)可知,pH值的變化對Cell@PEI的吸附效果影響顯著。當pH值<4.0時,p-ASA平衡吸附量及去除率隨pH值的升高而增大;當pH值=4.0時,p-ASA平衡吸附量及去除率最大;當pH值>4.0時,p-ASA的平衡吸附量和去除率呈明顯下降趨勢。由此可知,酸性條件有利于Cell@PEI對p-ASA的吸附,而堿性條件不利于吸附反應的進行。這是因為不同pH值下,p-ASA對應3個不同的pKa值(1.9、4.1和9.2)[33],即表面所帶電荷不同;酸性條件下,Cell@PEI在水溶液中可發生氨基表面質子化反應,使其表面帶正電荷,與帶負電荷的p-ASA發生靜電吸附,因而可提高其對p-ASA的去除率和平衡吸附量;堿性條件下,Cell@PEI表面攜帶的負電荷增多,與p-ASA之間形成的靜電斥力增大;另外,溶液中OH?增多也會對p-ASA的吸附造成一定的競爭效應,導致p-ASA的吸附受到抑制,因而去除率和平衡吸附量降低。綜上所述,溶液pH值會嚴重影響p-ASA的化學形態、溶解度、親水性等性能,最終影響其吸附特性。因此選取pH值=4.0為優選條件,此時p-ASA去除率為78.4%,Cell@PEI對p-ASA的平衡吸附量為98.0 mg/g。
2.2.2 吸附劑用量對Cell@PEI吸附性能的影響
當溶液pH值為4.0,p-ASA初始濃度為60 mg/L,吸附時間為250 min時,改變吸附劑用量,得到不同吸附量和去除率。圖6(b)為吸附劑用量對Cell@PEI吸附p-ASA的影響。從圖6(b)可知,隨吸附劑用量的增加,Cell@PEI對p-ASA的去除率逐漸增加,在吸附劑用量為0.07 g時達92.2%。這是因為溶液中吸附劑用量增加,p-ASA與吸附劑的接觸面積增大,吸附劑上活性位點增多;而平衡吸附量的降低可能是因為吸附劑用量的增加使Cell@PEI上的吸附位點呈不飽和狀態。

圖6 不同吸附條件對Cell@PEI吸附性能的影響Fig.6 Effects of different adsorption conditions on the adsorption perfomance of Cell@PEI
2.2.3p-ASA初始濃度對Cell@PEI吸附性能的影響
當吸附劑用量為0.02 g,吸附時間為250 min時,Cell@PEI對pH值為4.0的不同初始濃度下p-ASA的吸附性能如圖6(c)所示。由圖6(c)可知,隨著p-ASA初始濃度的增加(從10 mg/L提高至100 mg/L),平衡吸附量明顯提高,從6.78 mg/g提高至205.6 mg/g(從10 mg/L提高至100 mg/L)。這是因為當p-ASA初始濃度較低時,Cell@PEI表面的吸附位點多于p-ASA,此時平衡吸附量較低;而隨著p-ASA初始濃度的增加,吸附質和吸附劑的接觸機會增多,導致平衡吸附量增加。當p-ASA初始質量濃度增大時,其去除率整體呈先上升后略有降低的趨勢,并在25~100 mg/L范圍內趨于平衡。
2.2.4 共存離子對Cell@PEI吸附性能的影響
共存離子可與p-ASA競爭吸附劑上的吸附位點,本研究探究了3種常見的共存陰離子對Cell@PEI吸附p-ASA的影響,添加0.02 g Cell@PEI,控制p-ASA溶液pH值為4.0,初始濃度為60 mg/L,吸附時間為250 min,改變共存陰離子種類和濃度,進行吸附實驗,結果如圖6(d)所示。由圖6(d)可知,3種共存陰離子對Cell@PEI吸附p-ASA均有一定抑制作用,影響程度依次是:SO42?>CO32?>Cl?。隨著共存陰離子濃度的增加,抑制效果也愈加明顯,且SO42?比另2種陰離子對Cell@PEI吸附p-ASA的影響更大。這可能是由于氨基對SO42?有更高的親和力,使得SO42?可與p-ASA分子競爭活性吸附位點[34]。
2.2.5 吸附動力學分析
當吸附劑用量為0.02 g,對pH值為4.0、初始濃度為60 mg/L的p-ASA進行吸附實驗,探究吸附時間對Cell@PEI吸附p-ASA的影響,結果如圖7(a)所示。由圖7(a)可知,隨著吸附時間的延長,Cell@PEI吸附p-ASA的過程可分為:快速吸附過程(<60 min)和慢速吸附過程(>60 min)。Cell@PEI對于p-ASA的吸附主要集中在快速吸附過程,此時由于Cell@PEI表面具有豐富的活性吸附位點,吸附劑表面與溶液間有較大的濃度差,促使Cell@PEI吸附p-ASA,所以在此階段Cell@PEI對p-ASA的吸附速率較快,即其對p-ASA的去除快速增加。吸附60 min后,p-ASA的去除率和Cell@PEI的吸附量分別達80.9%和101.2 mg/g。隨著吸附反應的進行,Cell@PEI表面的有效活性位點隨之減少,固液界面濃度差下降,導致Cell@PEI對p-ASA的吸附速率下降,吸附反應趨于平衡。
將Cell@PEI對p-ASA的吸附量隨時間變化的數據用準一級動力學模型和準二級動力學模型進行擬合,結果如圖7(b)和圖7(c)所示。由圖7(b)和圖7(c)可以看出,準二級動力學模型的相關系數(R2)優于準一級動力學模型R2,說明準二級動力學模型能更好地描述Cell@PEI對p-ASA的吸附過程,表明吸附材料在吸附p-ASA的過程中,二者之間發生了靜電吸引和氫鍵連接,即Cell@PEI對p-ASA的吸附過程是化學吸附過程。
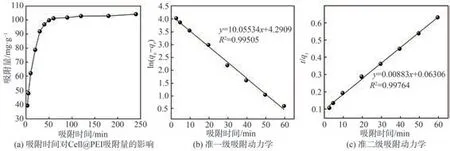
圖7 吸附動力學分析Fig.7 Adsorption kinetics analysis
2.2.6 吸附等溫線分析
為進一步研究Cell@PEI與p-ASA的相互作用機制,利用Langmuir和Freundlich模型分析其吸附過程,對不同溫度下的等溫吸附數據進行擬合,擬合結果如圖8所示。從圖8可以看出,Freundlich模型的相關系數(R2)高于Langmuir模型的R2,說明Freundlich吸附模型更適用于描述Cell@PEI對p-ASA的吸附過程,即Cell@PEI對p-ASA的吸附為非均勻、多分子層吸附。這可能是由于PEI作為官能化材料,為纖維素氣凝膠表面提供了更豐富的吸附作用力,快速吸附p-ASA至Cell@PEI表面后,吸附質進入氣凝膠三維結構內部再次擴散和物理吸附。表1和表2分別為Cell@PEI吸附p-ASA的Langmuir和Freundlich等溫吸附擬合參數。由表1和表2可知,Freundlich等溫式的吸附強度常數(1/n)小于1,Langmuir等溫式的RL在0~1之間,表明吸附容易進行。Langmuir等溫式和Freundlich等溫式的吸附平衡常數(KL、KF)及Qe均隨著溫度的升高而減小,表明低溫有利于Cell@PEI對p-ASA的吸附。
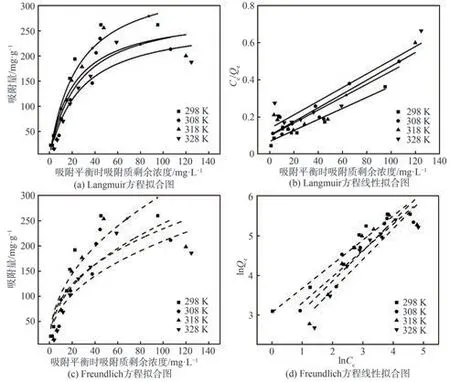
圖8 吸附等溫線分析Fig.8 Adsorption isotherm analysis
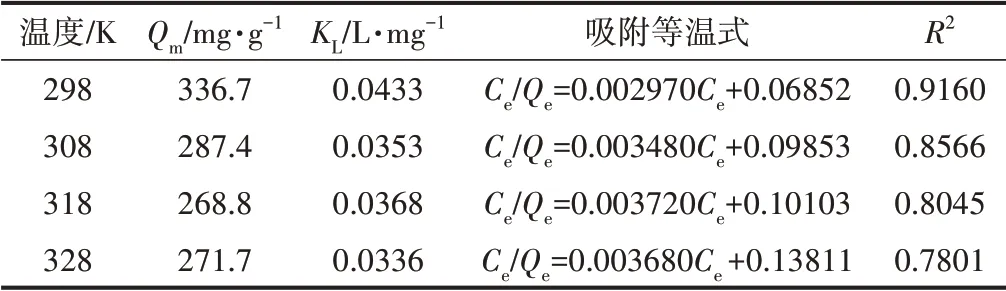
表1 Cell@PEI吸附p-ASA的Langmuir吸附等溫式Table 1 Langmuir adsorption isotherm for Cell@PEI towards p-ASA

表2 Cell@PEI吸附p-ASA的Freundlich吸附等溫式Table 2 Freundlich adsorption isotherm for Cell@PEI towards p-ASA
2.3 Cell@PEI吸附p-ASA的吸附機理研究
為了闡明p-ASA與Cell@PEI的分子間相互作用,本研究通過吸附前后Cell@PEI的XPS譜圖探究了p-ASA的吸附機理,結果如圖9所示。
2.3.1 氫鍵作用
分析FT-IR譜圖(見圖5)可知,Cell@PEI和p-ASA分子富含H—供體和H—受體基團(如C—H、—OH和N—H),這表明二者之間可以形成大量氫鍵。圖9(a)~(f)為吸附p-ASA前后Cell@PEI的各元素XPS譜圖。圖9(a)和圖9(d)分別為吸附p-ASA前后Cell@PEI C 1s譜圖,其中284.8、286.2和287.9 eV處的吸收峰分別對應Cell@PEI中的C—C/C==C、C—O/C==N和C==O的特征峰[35]。吸附p-ASA后,C—C/C==C占比由59.3%降至44.5%,C—O/C==N和C==O占比分別由36.3%和4.4%升至48.8%和6.7%,這可能是因為它們參與了氫鍵的形成。圖9(b)和圖9(e)顯示,將Cell@PEI的N 1s譜圖去卷積后得到2個峰,分別為399.0 eV的NH2/N—H和401.4 eV的NH3+[36],在吸附p-ASA后,NH2/N—H的占比由86.8%降至77.2%,而NH3+的占比由13.2%升至22.8%,這也表明了在吸附過程中氨基態發生了變化。從Cell@PEI的O 1s譜圖(見圖9(c)和圖9(f))可知,Cell@PEI中含有2種O峰,分別是532.3 eV的C—OH和531.2 eV的O…H[37],在吸附p-ASA后,C—OH的占比由70.4%升至88.7%,O…H的占比由29.6%降至11.3%,表明官能團C—OH參與了p-ASA吸附過程中O…H氫鍵的形成。因此,Cell@PEI與p-ASA之間存在氫鍵作用。
2.3.2 靜電引力
Cell@PEI對p-ASA的吸附是氣凝膠界面上發生的靜電相互作用的化學吸附過程。在吸附過程中p-ASA上具有較強還原活性的極性氨基,其偶極矩是由極性氨基的表面電荷分布不均勻所致。圖9(g)為不同pH值條件下Cell@PEI的Zeta電位圖。如圖9(g)所示,在pH值為2~10間,Cell@PEI Zata電位呈正電性。在pH值>4.0的條件下,p-ASA表面基本帶負電。而PEI是一種水溶性聚胺,在水中呈堿性,其分子鏈上擁有大量氨基N原子(伯、仲、叔胺的比例一般為1∶2∶1),具有很強的親質子性能[38]。結合pH值對吸附量的影響(詳見2.2.1),推測靜電作用是Cell@PEI吸附p-ASA過程的主要作用力之一。
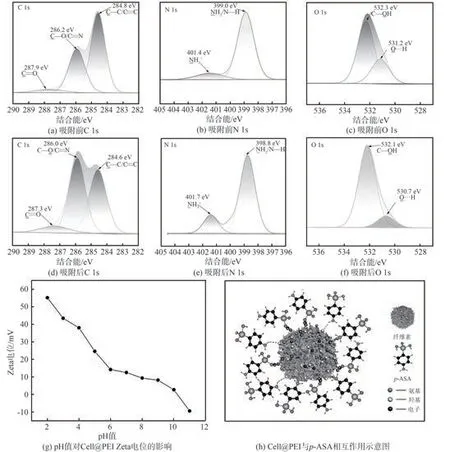
圖9 Cell@PEI吸附機理研究Fig.9 Adsorption mechanism analysis of Cell@PEI
2.4 再生使用性能分析
本研究分別選用0.2 mol/L和0.5 mol/L的NaOH溶液為解吸劑,對吸附p-ASA后的Cell@PEI進行解吸再生研究。圖10為不同濃度解吸劑對Cell@PEI進行5次解吸再生后的使用效果。由圖10所示,隨著解吸再生次數的增多,Cell@PEI對p-ASA的平衡吸附量和去除率均逐漸下降。5次解吸再生后,2種解吸液濃度下Cell@PEI對p-ASA的平衡吸附量分別保持在110.9 mg/g和105.4 mg/g,去除率分別達77.2%和73.4%。結果表明,Cell@PEI具有良好的可重復使用性和應用潛能。
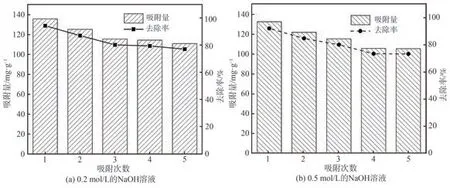
圖10 Cell@PEI解吸再生分析Fig.10 Analysis of desorption and regeneration of Cell@PEI
3 結論
本研究以纖維素為原料,利用聚乙烯亞胺(PEI)進行氨基改性,合成具有阿散酸(p-ASA)吸附功能的新型三維塊狀氨化纖維素氣凝膠(Cell@PEI),對Cell@PEI進行了理化性能分析及吸附機理探索。
3.1 在Cell@PEI對p-ASA的最佳吸附條件(p-ASA溶液pH值4.0,初始濃度為60 mg/L,吸附時間6 h,吸附溫度25℃,Cell@PEI添加量0.02 g)下,最大吸附量為205.6 mg/g。
3.2 Cell@PEI對p-ASA的吸附過程是化學吸附過程,符合準二級動力學方程;同時符合Freundlich等溫吸附方程,屬于非均勻、多分子層吸附,且吸附更易在低溫下進行。
3.3 吸附再生實驗結果表明,用2種不同濃度NaOH溶液循環再生5次后,Cell@PEI的平衡吸附量分別為110.9 mg/g和105.4 mg/g,去除率分別達77.2%和73.4%,表明Cell@PEI再生5次后仍可對水體中大部分p-ASA進行吸附,具有良好的再生使用效果。
