基于機器學習的分子動力軌跡預測
2023-03-13 15:24:10焦嘉
信息記錄材料 2023年1期
焦 嘉
(湖南信息職業技術學院 湖南 長沙 410203)
0 引言
分子動力模擬是一種計算分子和原子在相空間模擬運動的軌跡的計算機模擬方法。該方法主要利用計算機來計算模擬分子和原子在某段時間內運動結構變化[1],目前分子動力模擬發展已經比較成熟,能研究具有不同內部自由度的多原子分子、單原子分子,現在也已經成功運用在研究蛋白質、DNA等生物大分子的研究中[2-3]。當在各種條件下發生構象變化時,蛋白質構象在影響其功能中起著重要作用。分子動力學(molecular dynamics,MD)軌跡模擬可以提供蛋白質運動的結構數據,這在蛋白質功能中起著非常重要的作用。
現在比較流行的研究分子動力結構預測的方法是主成分分析(principal component analysis,PCA),在預測蛋白質結構中的用途,從而將前兩個(最高)主成分(PC)的線性組合用于生成新結構。與自動編碼器相比,對于更剛性的蛋白質,它的性能很好,但對于更柔性的蛋白質,效果不佳[4-6]。本文提出的方法主要就是解決對于柔性蛋白質的預測問題。
1 分子動力模擬結構的分類
本次實驗采用可視化分子動力學(visual molecular dynamics,VMD)軟件[7],該軟件能可視化呈現蛋白質的結構以及對不同軌跡的模擬分析。VMD顯示兩個軌跡圖像的總體視圖(見圖1),為了顯示構象差異,還繪制了兩個獨立軌跡的RMSD圖。
在圖1可視化了3D空間中的所有原子點后,根據繪制數據集在空間中的分布大致了解目標數據集的分布情況,確定數據集的分布,并根據數據的特征選擇合適的SVM分類器模型。
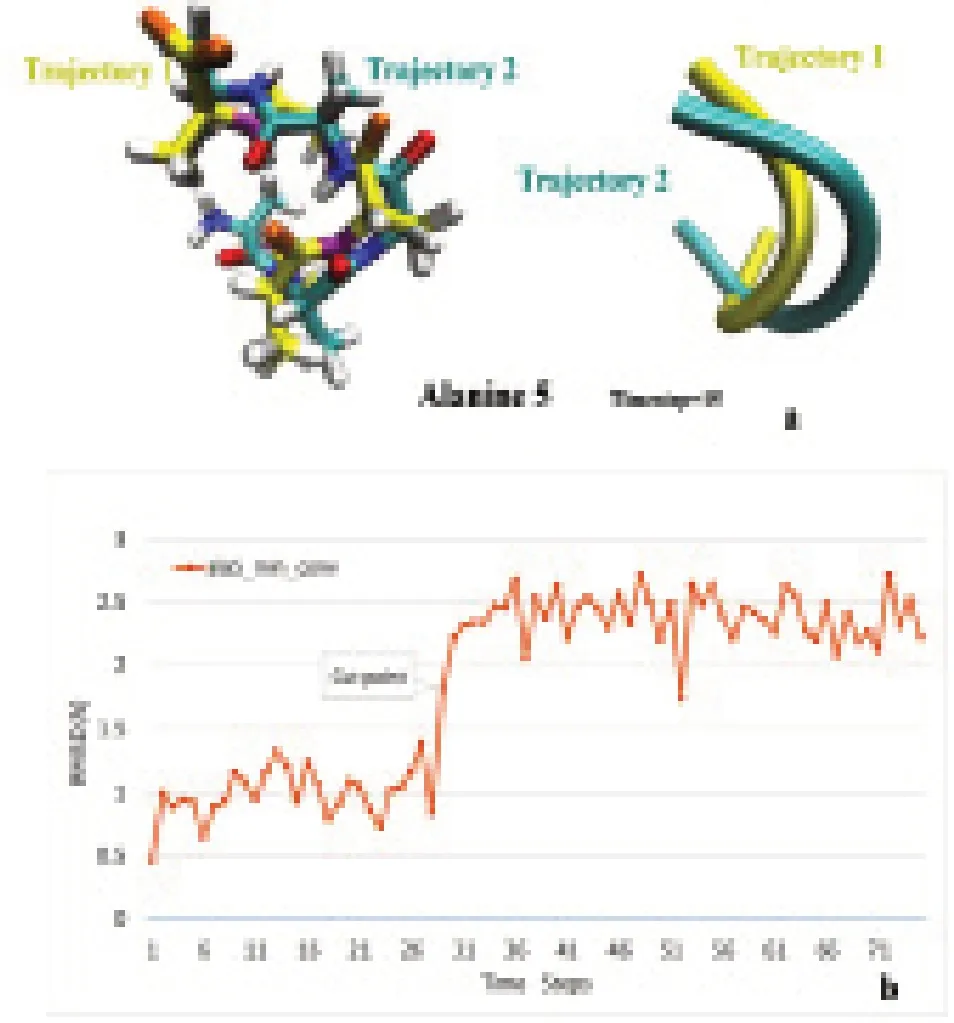
圖1 丙氨酸5的不同結構可視化示意圖和RMSD示意圖
2 分子動力模擬結構的異常檢測
2.1 一類支持向量機
在異常檢測的任務中,比較常用的就是一類支持向量機(one class support vector machine)[8-9],這個算法的原理就是把所有的訓練數據訓練出一個最小的超球面,這個超球面就盡量把所有訓練數據都“包”起來,當識別新的點的時候,如果落在超球面上,那么就是屬于這個類,反之則不是屬于這個類別。選擇One Class SVM(sklearn中的函數)的默認值RBF內核,以及gamma和nu超參數的默認值。另外,在這個實驗中,使用了Grid Search[10]來找到該模型的最佳參數對,并通過使用最佳組合超參數執行一類支持向量機的算法。圖2是一類支持向量機的原理圖,圖中黑點表示數據集,可以看到a、b兩個點離超球面的距離非常遠,所以這個算法把這兩個點視為異常點。

圖2 一類支持向量機原理示意圖
2.2 孤立森林
我們使用了另一種異常檢測方法來區分構象采樣中的差異——使用網格搜索通過準確性分數來搜索孤立森林(isolation forest)[11]的最佳組合。為了通過操縱isolation forest找出對性能的不同影響,還對每個超參數使用所有默認值對其進行訓練,并獲得在RMSD圖中顯示負點的結果。值得注意的是,將自動設置為污染時,參數行為不能“過時”。因此,將行為從“舊”更改為“新”。最重要的超參數是基本估計量、樣本數量和從該數據集中提取的特征,以及訓練模型中的估計量。就最大樣本數而言,為估計量和特征數設置了默認值,并從50、100、300到500中選擇了最大樣本數的值來訓練該模型。
2.3 離群點檢測法
離群點檢測法(local outlier factor, LOF)是一種基于距離的異常點檢測的算法,這個算法的主要思想是通過目標點T和其每個領域的點的密度來判斷被檢測點是否屬于異常,點T的密度越低就越可能被檢測成是異常點。這里的密度,指的是通過點之間的距離來計算的數值,樣本中點之間距離越遠,密度越低,距離越近,密度越高。同時因為LOF對密度的計算是通過點的第k鄰域來計算,而不是需要通過全局掃描式的計算,避免了時間復雜度很高的情況,這也是為什么叫“局部”異常因子算法的原因。下面是局部可達密度(local reachability density)的公式:
在公式(1)中,比值越接近1,說明p和周圍的點密度差不多,所以p可能是和周圍的點是同類。另外,如果比值小于1,則說明p是密集點,p大于1的話,說明p就是異常點。在異常檢測中,直接利用集成好的離群點檢測法算法在訓練集上,來對比前兩種算法的效果,最后得出對于該實驗最優的異常檢測算法。
3 分子動力模擬結構預測
3.1 改編的自編碼模型
這次實驗中,為了預測僅給出二維點的高維結構,我們提出了一種符合當前數據情況的編碼器模型,該編碼器模型通過輸入高維的坐標變量({x1,x2,x3…,xn}),轉化成二維的隱變量Z(恰好和輸入數據一樣的維度),然后再利用產生的二維擴展為高維的結構坐標作為解碼器(1,2,3。可以計算x和之間的誤差,通過控制兩編碼器和解碼器輸出的結果之間的誤差來訓練數據模型:
通過優化公式(2),最初訓練了一種具有較小誤差(損失)的改進型自動編碼器。完善的自動編碼器的損失函數的設計如圖3所示。其中x是輸入向量,x︿是輸出向量,盡量地使其與x非常相似,而z是隱向量,z'是新的隱向量對,這些參數通過最小化神經網絡中的值來訓練。

圖3 改編的自編碼器結構示意圖
改編的自動編碼器的思想來自另一個改編的自動編碼器,它是變分自動編碼器(VAE)[12-13],這是基于自動編碼器開發的神經網絡的另一種結構。它們之間的主要區別是損失函數,VAE使用Kullback-Leibler散度訓練了模型,從而最大限度地減少了編碼器分布和解碼器分布之間的損失。總的來說,這是一種有效的神經網絡結構,該結構以自我監督的方式進行訓練,可通過輸入大量結構坐標來近似重建幾何結構。另外,解碼器部分參考給定新的PC向量組(主要成分)的情況下產生新的預測結構的預測模型。通過查閱文獻,沒有論文研究過這種類型的自動編碼器,因此這是一種在傳統自動編碼器的基礎上進行修改的全新自動編碼器。
對于預測,我們就可以只利用經過訓練的自動編碼器中的解碼器部分,可以通過訓練相似的分布數據集(稱為生成模型)來泛化該模型,因此解碼器的輸出是針對不同數據的預測結果,但是這些數據都具有類似的分布和格式。對于任何潛在分布的采樣(PC對),預期此解碼器模型可以準確地重建原始輸入結構。在實際的實驗中,使用Keras在Python 3.8中構建改編的自動編碼器,Keras是具有Tensorflow的開源深度學習庫。本研究測試了具有3個編碼層和3個解碼層的網絡。在計算機科學方面,模型的損失反映了神經網絡的性能。 因此,比較了不同網絡產生的損耗結果,以評估結構的性能。圖4是整個模型的總體思路。
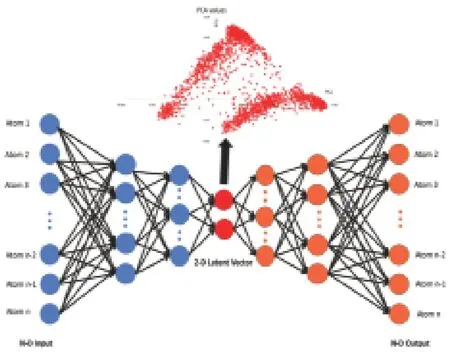
圖4 深度學習網絡訓練結構示意圖
3.2 改編的自編碼模型結果
本文選擇了訓練數據之外的12個點作為預測數據,以測試預測的準確性,為了測試改進的自動編碼器的性能和穩定性,在預測點周圍刪除了類似的結構(x的+/-0.002和y的+/-0.003之內的點,y與數據集中的x相比方差略大)。預測12點的每個點時的訓練數據集。例如,排除了正方形區域內的點。當預測(0,0.02)的結構時,訓練數據中的{(-0.002,0.02),(0.002,0.02),(0,0.017),(0,0.023)}。在劃分2 000條軌跡時,仍然采用相同的比率(訓練數據為80%,測試數據為20%),并對它們進行了相同的MinMax歸一化處理。實驗結果如圖5所示。
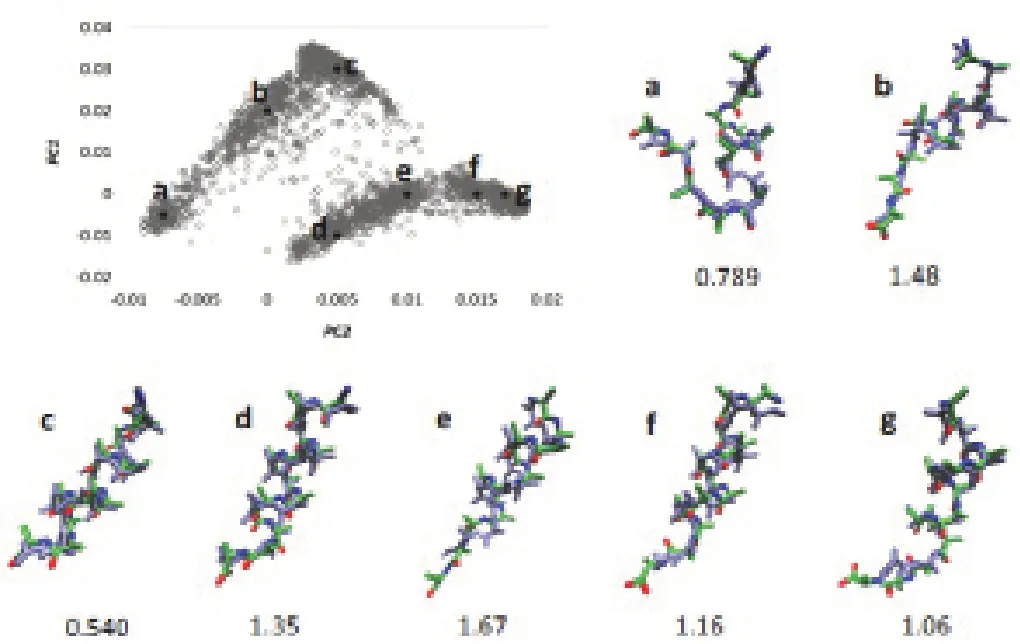
圖5 預測結構和真實結構對比圖
圖5左上方:代表了2D RMSD矩陣(灰色圓圈)的PC1與PC2圖,點a~g(黑點)用于測試算法。對于每個預測,將(PC1,PC2)值在(+/-0.002,+/-0.003)以內的點從訓練集中排除。對于每個點(a~g),都顯示了具有預測結構的原始結構的疊加圖,下面的數值代表了其RMSD值。
4 結語
本文通過計算機領域的機器學習和深度學習的使用,在分子動力模擬領域的一系列探究實驗,充分證明了機器學習和深度學習在該領域的可行性和可探究性。由于之前很少有人在這個領域用機器學習的方法來對分子運動軌跡進行研究或者預測,之前的一些研究主要是利用類似于PCA的降維方法來觀察分子動力模擬軌跡的分布,但是并不能很準確地預測出分子結構,這些問題其實一直困擾著這個領域,所以現在用機器學習和深度學習的方法來進行這些探究實驗的意義非常重大,為該領域開辟了一個全新的研究視角。本文提出了運用機器學習和深度學習的方法來對分子動力模擬領域的原子坐標進行探究,成功驗證了機器學習算法在蛋白質軌跡分類的實驗上的有效性,用三種異常檢測算法對不同狀態的蛋白質大分子結構進行檢測,通過對比不同RMSD值的變化,來判斷具體時刻的分子結構變化的程度,從而得到“異常”的蛋白質分子結構,最后提出了一種全新的基于自編碼的神經網絡模型,通過修改損失函數的組成來構建適合低維輸入和高維輸出情況的模型。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
光學精密工程(2016年6期)2016-11-07 09:07:19
核科學與工程(2015年4期)2015-09-26 11:59:03
現代企業(2015年9期)2015-02-28 18:56:50
