EGFR酪氨酸激酶及其抑制劑的研究進(jìn)展
2023-07-21 01:15:14朱美靈夏德斌
黑龍江大學(xué)自然科學(xué)學(xué)報(bào) 2023年3期
關(guān)鍵詞:耐藥
關(guān) 靜, 朱美靈, 夏德斌
(1.哈爾濱醫(yī)科大學(xué) 藥學(xué)院, 哈爾濱 150081; 2.哈爾濱工業(yè)大學(xué) 化工與化學(xué)學(xué)院, 哈爾濱 150001)
0 引 言
EGFR是原癌基因c-erbB1的表達(dá)產(chǎn)物,是人表皮生長(zhǎng)因子受體(Human epidermal growth factor receptor, HER)家族成員之一。研究發(fā)現(xiàn),EGFR分為三區(qū):胞外配體結(jié)合區(qū)、跨膜區(qū)和胞內(nèi)激酶區(qū),廣泛分布于各種上皮細(xì)胞的細(xì)胞膜上。EGFR基因位于第七號(hào)染色體上,由118個(gè)堿基組成,包括28個(gè)外顯子。其轉(zhuǎn)錄形成的mRNA長(zhǎng)約5.6 kb,編碼的EGFR是分子量為170 kD的跨膜糖蛋白。編碼蛋白由1 186個(gè)氨基酸組成,具有氨基酸激酶(Tyrosine kinase, TK)活性,可以將胞內(nèi)信號(hào)傳遞到胞外[1]。已報(bào)道的EGFR配體有表皮生長(zhǎng)因子EGF、結(jié)合肝素的EGF、轉(zhuǎn)化生長(zhǎng)因子TGF、β-細(xì)胞素和雙調(diào)蛋白等[2]。EGFR信號(hào)通路過度活化和EGFR自身過度表達(dá)是惡性腫瘤的主要發(fā)生機(jī)制[3-4]。EGFR相關(guān)信號(hào)通路主要有RAS/MAPK、NF-KB、PI3K/AKT/mTOR和JAK/STAT等[5]。與配體結(jié)合后,EGFR由單體轉(zhuǎn)化為二聚體,然后激活它位于細(xì)胞內(nèi)的激酶通路,包括Y992、Y1045和Y1148等激活位點(diǎn),使下游磷酸化,誘導(dǎo)細(xì)胞增殖,腫瘤生長(zhǎng)。而在實(shí)體腫瘤中EGFR高表達(dá)或異常表達(dá),研究表明,EGFR基因突變和蛋白的過度表達(dá)都可以激活下游信號(hào)通路[6],這與癌癥密切相關(guān)。
主要的EGFR靶向藥物可以分為兩大類:一類是單克隆抗體(簡(jiǎn)稱單抗),作用于胞外區(qū),競(jìng)爭(zhēng)性地抑制配體與EGFR的結(jié)合,從而阻斷EGFR酪氨酸激酶的活化;另一類是小分子酪氨酸激酶抑制劑(Tyrosine kinase inhibitors, TKIs),它進(jìn)入細(xì)胞內(nèi),干擾三磷酸腺苷(Adenosine-triphosphate, ATP)結(jié)合,抑制21 bp氨基酸激酶磷酸化。雖然EGFR靶向治療已成功應(yīng)用于臨床并取得了一定的療效,但治療過程中患者出現(xiàn)的耐藥現(xiàn)象(包括原發(fā)性耐藥和獲得性耐藥兩種)仍是一個(gè)亟待攻克的難題。發(fā)展和創(chuàng)新EGFR抑制劑是一個(gè)重要的研究目標(biāo),本文對(duì)TKIs代表藥物進(jìn)行綜述。
1 EGFR-TKIs的研發(fā)進(jìn)展
惡性腫瘤是一類嚴(yán)重危害人類健康的疾病,研究和開發(fā)高效、選擇性強(qiáng)的低毒抗腫瘤藥物是藥學(xué)工作者的目標(biāo)之一。肺癌是世界上最常見的癌癥,每年都有超過100萬人死于肺癌,而80%~90%患者為非小細(xì)胞肺癌(Non small-cell lung cancer, NSCLC)[7]。在非小細(xì)胞肺癌中,EGFR突變大約占12%~47%,其中最為常見的兩種突變是19號(hào)外顯子的缺失突變(del19)和21號(hào)外顯子的點(diǎn)突變(主要是L858R)[8]。EGFR是癌癥生長(zhǎng)的刺激因素,與腫瘤的發(fā)生有密切關(guān)系,EGFR-TKIs成為抗腫瘤藥物研發(fā)的重要方向。
吉非替尼(Gefitinib, ZD1839)和厄洛替尼(Erlotinib, OSI774)是第一批用于治療非小細(xì)胞肺癌的EGFR-TKIs,并取得一定療效,兩者對(duì)野生型EGFR有效,對(duì)L858R和delE746-A750有敏感性[9]。Gefitinib是一種口服型、苯胺喹唑啉類可逆性酪氨酸激酶抑制劑。吉非替尼在聯(lián)合化療中取得明顯效果,它在與順鉑、卡鉑、奧沙利鉑和紫杉醇等細(xì)胞毒性藥物聯(lián)合用藥時(shí)能增強(qiáng)抑制作用,促進(jìn)腫瘤細(xì)胞凋亡[10]。厄洛替尼(Eelotiinib, OSI774)是第一個(gè)獲得美國食品公司藥品管理局批準(zhǔn)的一類口服型EGFR-TKIs藥物,與化療類藥物聯(lián)合使用可以獲得更好的治療效果和生存期,改善患者生存質(zhì)量[11]。然而,患者對(duì)這些抑制劑在9~13個(gè)月后產(chǎn)生獲得性耐藥。常見的與耐藥相關(guān)的EGFR突變有19號(hào)外顯子缺失、L858R突變和T790M突變,而大約50%~60%的耐藥是由于EGFR T790M突變[12]。另外,埃克替尼(Icotinib, BPI-2009)于2011年6月7日獲中國食品藥品監(jiān)督管理總局批準(zhǔn)上市,打破了非小細(xì)胞肺癌,特別是晚期非小細(xì)胞肺癌治療由進(jìn)口藥品壟斷的格局。埃克替尼對(duì)晚期NSCLC的療效與吉非替尼相當(dāng),安全性較吉非替尼更優(yōu),中位無進(jìn)展生存期為4.6個(gè)月。TKIs靶向藥物對(duì)血腦屏障的穿透率分別為埃克替尼 6.1%,厄洛替尼2%~4%,吉非替尼1%,因此對(duì)于存在腦轉(zhuǎn)移和21號(hào)外顯子突變的患者,埃克替尼是第一代TKIs藥物中的首選用藥。
德國勃林格殷格翰公司研發(fā)的第二代EGFR-TKIs藥物阿法替尼(Afatinib, BIBW2992)是一種新型口服制劑,是EGFR/HER2雙重酪氨酸激酶受體的不可逆抑制劑。它與具有催化作用的EGFR第773位酪氨酸結(jié)合,以非ATP競(jìng)爭(zhēng)方式使受體蛋白不可逆改變,對(duì)EGFR野生型和突變型包括T790M都有很好的抑制作用[12]。阿法替尼與T790M突變的結(jié)合能力是第一代TKIs的100倍以上。阿法替尼與西妥昔單抗聯(lián)合治療可有效抑制T790M耐藥突變的NSCLC原代細(xì)胞[14]。針對(duì)EGFR激活突變的患者,通過阿法替尼和順鉑分別聯(lián)用培美曲塞的對(duì)比,可觀察到阿法替尼與培美曲塞聯(lián)用對(duì)T790M突變的NSCLC存在明顯的協(xié)同生長(zhǎng)抑制作用,尤其是對(duì)T790M突變可逆性EGTR-TKIs耐藥患者[15]。但是阿法替尼對(duì)野生型EGFR和突變型EGFRs有相同的結(jié)合力,缺失了對(duì)野生型EGFR的選擇性,導(dǎo)致產(chǎn)生毒性和較嚴(yán)重的副作用[16]。達(dá)克替尼(Dacomitinib, PF299804)是美國輝瑞公司研制的第二代、不可逆的EGFR酪氨酸激酶抑制劑,該藥作用機(jī)制類似阿法替尼,能不可逆抑制三種不同ERBB家族分子成員,包括EGFR(HER1)、HER2和HER4。達(dá)克替尼用于攜帶EGFR激活突變的局部晚期或轉(zhuǎn)移性NSCLC患者的一線治療,死亡或疾病進(jìn)展風(fēng)險(xiǎn)顯著降低了41%,中位無進(jìn)展生存期為14.7個(gè)月,總緩解率為75%,中位緩解持續(xù)時(shí)間為14.8個(gè)月[17]。
通常在服用一代或二代EGFR抑制劑后9.2~14.7個(gè)月時(shí),患者會(huì)出現(xiàn)不同程度的耐藥性。在這些耐藥突變中,約有50%~70%發(fā)生在EGFR上門控位置(Gatekeeper)T790M。該突變可引起EGFR空間構(gòu)像改變,增加EGFR對(duì)ATP的親和力,從而削弱抑制劑與EGFR的結(jié)合能力。二代EGFR抑制劑阿法替尼雖然在體外對(duì)EGFR-T790M突變有抑制活性,但在臨床應(yīng)用中仍然未能克服T790M突變產(chǎn)生的耐藥性。且一代和二代EGFR抑制劑都很難排除對(duì)野生型EGFR的抑制,從而導(dǎo)致明顯的皮膚毒性(如痤瘡樣皮疹)。直到第三代抑制劑奧西替尼出現(xiàn),這種情況才得以解決,如圖1所示。奧西替尼(Osimertinib, AZD9219)和奧母替尼(Olmutinib, HM61713)是針對(duì)T790M突變的第三代EGFR-TKIs,對(duì)EGFR敏感突變和T790M耐藥突變有更好的療效。奧西替尼為單苯胺基嘧啶環(huán)化合物,通過與EGFR激酶區(qū)ATP結(jié)合域的半胱氨基-797殘基不可逆共價(jià)結(jié)合來抑制EGFR活性[18]。將奧西替尼單藥用于一線和二線治療,療效良好,具有安全性和可耐受性。Janne等對(duì)局部進(jìn)展或轉(zhuǎn)移的EGFR-TKIs耐藥患者的治療效果、不良反應(yīng)藥效量、有效性及安全性進(jìn)行了評(píng)估[19-21]。而對(duì)于聯(lián)合治療,體外研究結(jié)果顯示,如果奧西替尼與 MEK阻滯劑司美替尼和曲美替尼聯(lián)合使用能有效延遲耐藥細(xì)胞的生長(zhǎng),意味著奧西替尼與其他抗腫瘤藥物聯(lián)用也許可延緩?qiáng)W西替尼耐藥的發(fā)生[22-23]。奧母替尼也是針對(duì)T790M突變的第三代EGFR-TKIs,在2016年于韓國批準(zhǔn)上市,用于既往接受過TKIs治療的局部晚期或轉(zhuǎn)移性EGFRT790M突變NSCLC患者。然而治療病人很高概率發(fā)生了第三代的C797S突變,其他突變還有:EGFR L718Q突變、L718Q.L844V和C797S突變,HER2和MET擴(kuò)增等,使第三代EGFR-TKIs出現(xiàn)了耐藥抵抗[24]。EGFR 與 VEGFR均可通過激活下游信號(hào)通路PI3K/AKT和Ras/Raf/Erk影響細(xì)胞生理功能。在EGFR突變的NSCLC中,上調(diào)的 EGFR 突變通過不依賴缺氧的機(jī)制增加VEGF,進(jìn)而促進(jìn)了對(duì)EGFR-TKIs耐藥的產(chǎn)生[25]。
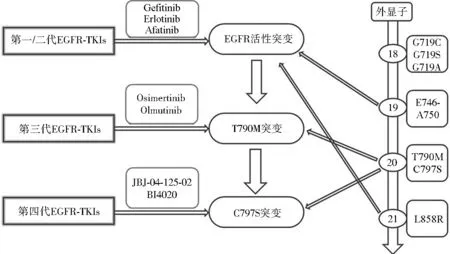
圖1 EGFR突變及EGFR-TKIs代表藥物
EAI045是第一個(gè)針對(duì)T790M和C797sEGFR突變的變構(gòu)非ATP競(jìng)爭(zhēng)性抑制劑。研究者證明了EAI045能顯著提高二聚體缺陷的EGFR突變活性,其與西妥昔單抗聯(lián)合使用能顯著抑制具有L858R/T790M突變的Ba/F3細(xì)胞增殖。因?yàn)樗臍埢h(yuǎn)離變構(gòu)結(jié)合口袋,C797S突變不會(huì)影響EAI045的療效,但此變構(gòu)抑制劑單獨(dú)使用無效,聯(lián)合使用西妥昔單抗可使EAI045抵制T790M和C797s活性充分提高。西妥昔單抗不是EGFR突變體特異性的單克隆抗體,可能導(dǎo)致靶向WT EGFR相關(guān)毒性,所以EAI045未能進(jìn)入臨床試驗(yàn)[26-27]。
2019年,丹娜-法伯癌癥研究院研發(fā)的JBJ-04-125-02在體外和體內(nèi)均可抑制細(xì)胞增殖和EGFRL858R/T790M/C797S信號(hào)傳導(dǎo),而且毒性比EAI045小很多,但JBJ-04-125-02對(duì)EGFRDel19/T790M/C797S突變無效。EGFR與其配體結(jié)合后,在細(xì)胞表面形成二聚體,細(xì)胞內(nèi)自磷酸化后激活下游傳導(dǎo)通路。JBJ-04-125-02對(duì)EGFR二聚體含量高的腫瘤細(xì)胞株抑制能力弱,而西妥昔單抗能夠阻斷EGFR二聚體的形成,JBJ-04-125-02聯(lián)合西妥昔單抗增強(qiáng)了抗腫瘤活性。而且,奧希替尼同樣可以增強(qiáng)JBJ-04-125-02的抗腫瘤活性,且副作用比西妥昔單抗方案小。2019年底,該研究院對(duì)JBJ-04-125-02聯(lián)合奧希替尼開啟了一期臨床實(shí)驗(yàn)[28]。
2019年10月,我國正大天晴藥業(yè)集團(tuán)研發(fā)的四代EGFR靶向藥TQB3804正式步入I期臨床實(shí)驗(yàn)。TQB3804不但可以解決奧西替尼的耐藥問題,而且能抑制1/2代TKIs耐藥后的T790M雙重突變,具體如表1所示。

表1 EGFR-TKIs代表藥物的基本信息
2019年11月,勃林格殷格翰制藥公司的科學(xué)家通過高通量篩選和基于結(jié)構(gòu)的藥物設(shè)計(jì)研發(fā)了第四代EGFR抑制劑BI-4020。蛋白及細(xì)胞實(shí)驗(yàn)均顯示,其能有效抑制T790M和/或C797S突變的EGFR,并在含順式三突變del19/T790M/C797S的小鼠異體移植NSCLC腫瘤模型(Human PC-9)上顯示很好的藥效[29]。
2020年1月,韓國的Bridge Biotherapeutics 公司宣布,美國食品和藥物管理局批準(zhǔn)了BBT-176作為EGFR靶向的酪氨酸酶抑制劑治療非小細(xì)胞肺癌進(jìn)入臨床實(shí)驗(yàn)。
2 EGFR-TKIs與EGFR蛋白的相互作用關(guān)系
2.1 第一代EGFR-TKIs
EGFR分子的第790位殘基處在非常重要的位置,它位于蛋白結(jié)構(gòu)的連接N和C-lobe的鉸鏈部分,并且處于ATP結(jié)合口袋深處, 可以使吉非替尼和厄洛替尼分子與蛋白充分靠近。因此,門控位置 (Gatekeeper)的殘基是一個(gè)激酶與小分子藥物結(jié)合的特異性的重要決定因素。吉非替尼和厄洛替尼具有4-苯胺喹唑啉的內(nèi)核,晶體學(xué)結(jié)構(gòu)提示,兩種藥物對(duì)于野生型或突變體激酶結(jié)合方式相似,通過與ATP競(jìng)爭(zhēng)位于激酶區(qū)的ATP結(jié)合位點(diǎn),抑制EGFR的酪氨酸激酶活性,如圖2和圖3所示。發(fā)生T790M突變后,蛋氨酸(Met)側(cè)鏈變長(zhǎng),導(dǎo)致了第一代EGFR-TKIs與ATP口袋結(jié)合的位阻性,阻斷與藥物的最佳相互作用,同時(shí)與ATP的親和力高于EGFR-TKIs,從而使ATP競(jìng)爭(zhēng)抑制劑失效[30-32]。

圖2 吉非替尼的結(jié)構(gòu)及其與EGFR蛋白激酶結(jié)構(gòu)域的鍵合模式

圖3 厄洛替尼的結(jié)構(gòu)及其與EGFR蛋白激酶結(jié)構(gòu)域的鍵合模式
2.2 第二代EGFR-TKIs
阿法替尼的喹唑啉環(huán)是重要的結(jié)構(gòu)單元,與ATP競(jìng)爭(zhēng)細(xì)胞內(nèi)的激酶結(jié)構(gòu)域,從而阻礙下游信號(hào)的傳導(dǎo)。 喹唑啉母核上的N-1原子與激酶Met793上的NH形成分子間氫鍵。喹唑啉環(huán)C4位上的2-氯-3氟苯胺基與空間上方Val726和Met766的分子間作用力使阿法替尼進(jìn)一步深入ATP結(jié)合位點(diǎn)。喹唑啉母核C6位上側(cè)鏈的烯丙酰結(jié)構(gòu)對(duì)Afatinib發(fā)揮抗腫瘤活性作用至關(guān)重要,它與EGFR上半胱氨酸殘基Cys797的巰基發(fā)生邁克爾加成反應(yīng),可使激酶失活,不可逆地抑制酪氨酸激酶的活性,如圖4所示[33-34]。研發(fā)人員通過結(jié)構(gòu)鑒定進(jìn)一步證明了共價(jià)鍵的存在,并且發(fā)現(xiàn)阿法替尼通過與HER2的Cys805以及HER4的Cys803相互作用而抑制酶的活性[35]。

圖4 阿法替尼的結(jié)構(gòu)及其與EGFR蛋白激酶結(jié)構(gòu)域的鍵合模式
2.3 第三代EGFR-TKIs
奧西替尼和奧母替尼均具有一個(gè)“U”型結(jié)構(gòu),有利于與表皮因子受體目標(biāo)結(jié)合。第三代EGFR-TKIs有丙烯酰胺的結(jié)構(gòu)單元,它可以作為受體與在表皮因子受體ATP結(jié)合域內(nèi)的Cys797的活性巰基發(fā)生邁克爾加成反應(yīng)形成共價(jià)鍵,而帶有C797S突變的EGFR不能形成共價(jià)鍵。因?yàn)樵谏項(xiàng)l件下,絲氨酸的無活性巰基不能形成共價(jià)鍵,提供一個(gè)關(guān)鍵機(jī)制的阻力[9]。在晶體結(jié)構(gòu)中,奧西替尼結(jié)合在P-loop和Pro794、Cys797之間的蛋白質(zhì)骨架的ATP結(jié)合口袋外邊緣。有兩個(gè)氫鍵幫助錨定配體,即奧西替尼的N4氫原子與Met793的N原子;羰基氧原子與Cys797的N原子結(jié)合形成兩個(gè)氫鍵幫助固定配體。奧西替尼的取代苯環(huán),與下面的蛋白質(zhì)骨架發(fā)生范德華相互作用,與上面的非極性殘基廣泛作用,完成了夾心效果,Leu718與奧西替尼的苯環(huán)接觸,Ala743與嘧啶環(huán)接觸,Val726和Phe723與吲哚環(huán)接觸,Leu718-Gly719的蛋白質(zhì)骨架也與吲哚環(huán)的邊緣形成范德華相互作用力,如圖5所示[36]。

圖5 奧西替尼的結(jié)構(gòu)及其與EGFR蛋白激酶結(jié)構(gòu)域的鍵合模式
同為第三代EGFR-TKIS的還有艾氟替尼(Alflutinib, AST2818),它是上海愛利斯特制藥有限公司開發(fā)的一種基于三氟乙氧基吡啶的不可逆EGFR-TKIs,對(duì)EGFR耐藥突變(如G719X、19號(hào)外顯子缺失、L858R、L861Q和T790M)具有較高的選擇性,對(duì)野生型EGFR無抑制作用。
2.4 第四代EGFR-TKIs
奧西替尼雖然解決了T790M突變的問題,但臨床上已經(jīng)觀察到,進(jìn)行奧西替尼二線治療的EGFRT790M陽性NSCLC患者,服藥10個(gè)月后出現(xiàn)耐藥現(xiàn)象,其中20%~40%為C797S突變(包含del19/T790M/C797S或L858R/T790M/C797S的順式或反式三突變)。因此,開發(fā)下一代EGFR抑制劑需要滿足更多臨床需求:一是對(duì)del19突變(最常見的突變形式)的EGFR具有高活性;二是能解決耐藥突變T790M和/或C797S;三是對(duì)野生型EGFR無作用;四是在整個(gè)人類激酶組顯示高選擇性。EAI045與EGFR作用呈“Y”形或“三葉螺旋槳”形,從EGFRT790M突變激酶的共晶結(jié)構(gòu)可以看出,抑制劑EAI001與經(jīng)典的ATP結(jié)合口袋附近的變構(gòu)口袋結(jié)合,部分通過由內(nèi)向外形成αC-螺旋結(jié)構(gòu),結(jié)合后作為一個(gè)“三葉螺旋槳”或“Y”形配置。氨噻唑片段延伸至T790M守門人蛋氨酸與Lys745殘基活性位點(diǎn)之間,視為T790M突變體的選擇性;苯基延伸到ATP結(jié)合口袋后面的疏水空隙,與Leu-777和Phe-856殘基相互作用,而第三個(gè)區(qū)域,1-氧異吲哚基沿αC-螺旋結(jié)構(gòu)向溶液暴露區(qū)延伸。另外,EAI001氨基組在Asp-Phe-Gly(DFG)序列與Asp-855之間形成氫鍵。進(jìn)一步藥物化學(xué)優(yōu)化使苯基組產(chǎn)生了更大的影響力,也就是EAI045對(duì)野生型EGFR保持千倍的選擇性。然而EAI045的結(jié)合位點(diǎn)是EGFR激酶在不活躍的構(gòu)象時(shí)由αC-螺旋運(yùn)動(dòng)產(chǎn)生的。兩個(gè)亞基的不對(duì)稱二聚體構(gòu)成的異構(gòu)結(jié)合位點(diǎn)不可接近,因此單獨(dú)使用EAI045的療效是有限的。當(dāng)與西妥昔單抗聯(lián)合使用時(shí),EAI045能成功到達(dá)每一種EGGFR激酶結(jié)構(gòu)域[37-38],EAI045的噻唑、異吲哚啉-1-酮和對(duì)氟苯酚的芳香環(huán),與EGFR激酶的多個(gè)疏水基團(tuán)進(jìn)行交互作用。具體來說,噻唑基團(tuán)與Met790側(cè)鏈相互作用,對(duì)氟苯酚的苯基與Phe856側(cè)鏈相互作用,異吲哚啉-1-酮基團(tuán)與Lle759、Leu747、Leu788、Leu777和Met766側(cè)鏈發(fā)生廣泛的疏水性相互作用,如圖6所示。極性在EAI045與EGFR激酶的相互作用中起到了重要作用。化合物的酰胺氮原子與EGFR的Asp855主鏈羧基形成氫鍵,異吲哚啉-1-酮的羰基氧與Lys745的側(cè)鏈ε-胺基形成另一個(gè)氫鍵[39-40]。

圖6 EAI045的結(jié)構(gòu)及其與EGFR蛋白激酶結(jié)構(gòu)域的鍵合模式
另外,吡啶并[3,4-d]嘧啶類化合物可以作為新的EGFR-TKIs[41-42]。吡啶并[3,4-d]的7位氮原子和6位的氨基吡啶氫原子,在鉸鏈區(qū)域與Met793形成了兩個(gè)氫鍵,支架2位的苯胺氫原子與看門人殘基Met790形成氫鍵,4位的氨基吡啶或氨基哌啶占據(jù)ATP核糖袋,并從Asp-Phe-Gly序列與Phe856和Asp產(chǎn)生范德華相互作用。位于吡啶并[3,4-d]嘧啶2位的苯胺插入背側(cè)疏水袋(BHP)產(chǎn)生疏水作用,這個(gè)疏水作用是必要的。4位的氨基哌啶上含有羥基,能使突變Ser797的氫鍵作用增強(qiáng),能夠被設(shè)計(jì)成針對(duì)Ser797突變的小分子抑制劑。
JBJ-04-125-02的結(jié)構(gòu)式如圖7(a)所示,它能夠與EGFR的變構(gòu)袋結(jié)合,這種變構(gòu)袋是由失活激酶構(gòu)象中輪狀核c-螺旋向外位移產(chǎn)生的,如圖7(b)所示。噻唑酰胺、苯環(huán)和異吲哚啉酮的結(jié)合模式與之前觀察到的EAI001相似[40]。此外,在DFG基序中,羥基與Phe856的羰基形成氫鍵。4-哌嗪基苯基沿著環(huán)-螺旋延伸到溶劑暴露外表面,其苯環(huán)在激酶p-環(huán)上與Phe723發(fā)生π-π堆積。有趣的是,該化合物的結(jié)合誘導(dǎo)了激酶激活環(huán)的新構(gòu)象,激活環(huán)中哌嗪和Glu865之間的氫鍵穩(wěn)定了該構(gòu)象,如圖7(c)所示。此外,Glu749定位在哌嗪基團(tuán)的氫鍵上。預(yù)計(jì)與EAI045相比,重新配置的活化環(huán)和與哌嗪基團(tuán)形成的氫鍵有助于增強(qiáng)該化合物的效力[28]。

圖7 (a) JBJ-04-125-02化學(xué)結(jié)構(gòu)式; (b)與JBJ-04-125-02和AMP-PNP結(jié)合的 EGFRT790M/V948R晶體結(jié)構(gòu); (c)變構(gòu)結(jié)合口袋
3 展 望
隨著醫(yī)療技術(shù)的不斷發(fā)展,人們對(duì)腫瘤的致病機(jī)制進(jìn)一步了解,靶向治療成為研究熱點(diǎn)。不斷發(fā)展的靶向藥物能夠抑制酪氨酸激酶在細(xì)胞內(nèi)的信號(hào)傳導(dǎo),在腫瘤發(fā)生侵襲時(shí)發(fā)揮重要作用,針對(duì)EGFR-TKIs開發(fā)的一系列分子靶向抑制劑將會(huì)為腫瘤患者帶來希望。另外,隨著血液樣本中ctDNA檢測(cè)技術(shù)的快速發(fā)展,癌癥個(gè)體化深度測(cè)序分析方法(Cancer personalized profiling by deep sequencing, CAPP-Seq)已經(jīng)能夠準(zhǔn)確測(cè)定非小細(xì)胞肺癌突變基因的分子分型,指導(dǎo)EGFR-TKIs靶向藥物的選用。未來基因檢測(cè)技術(shù)將適用于多種類型的惡性腫瘤并廣泛應(yīng)用于臨床,相信會(huì)有更多療效精準(zhǔn)的靶向藥物問世,促進(jìn)個(gè)體化癌癥治療的發(fā)展,改善人類的健康狀態(tài)。
猜你喜歡
保健醫(yī)苑(2022年5期)2022-06-10 07:46:38
現(xiàn)代臨床醫(yī)學(xué)(2022年3期)2022-06-06 07:59:40
昆明醫(yī)科大學(xué)學(xué)報(bào)(2022年1期)2022-02-28 07:43:40
天津醫(yī)科大學(xué)學(xué)報(bào)(2021年3期)2021-07-21 09:04:02
科學(xué)大眾(2020年12期)2020-08-13 03:22:22
云南醫(yī)藥(2019年3期)2019-07-25 07:25:10
現(xiàn)代檢驗(yàn)醫(yī)學(xué)雜志(2016年1期)2016-11-12 13:19:40
國外醫(yī)藥(抗生素分冊(cè))(2016年6期)2016-07-10 11:34:45
中國衛(wèi)生標(biāo)準(zhǔn)管理(2015年14期)2016-01-15 02:58:37
中國當(dāng)代醫(yī)藥(2015年17期)2015-03-01 02:03:58
