基于HIF-1信號通路基因變化探討COPD肺血管病變的發生機制
2023-07-26 10:38:12李小娟郭思佳孫增濤王坤李曉丹岳寶柱欒哲宇
中國老年學雜志 2023年14期
關鍵詞:模型
李小娟 郭思佳 孫增濤 王坤 李曉丹 岳寶柱 欒哲宇
(1天津中醫藥大學第二附屬醫院呼吸科,天津 300250;2天津中醫藥大學;3鄭州市第三人民醫院呼吸科)
慢性阻塞性肺疾病(COPD)是一種以氣流受限為主要特征的呼吸系統常見病與高發病。該病雖可預防和治療,但由于病程遷延反復,疾病往往呈進行性發展,嚴重影響患者的生活質量和勞動能力,為社會帶來沉重負擔〔1〕。據統計,我國目前COPD患者已達到約1億人,其中40歲以上人群的患病率高達13.7%〔2〕。COPD患者的肺血管病變是其特征性病理學改變之一,也是其發展成肺心病的重要原因。研究顯示,COPD患者中每年有約6%發展至肺心病,尸檢資料顯示,COPD患者中約有40%存在右心室肥大〔3〕。因此,認識COPD進展中肺血管病變的形成機制,針對性進行干預,是延緩COPD發展為肺心病的關鍵。
低氧誘導因子(HIF)-1是機體中調節氧穩態的核心轉錄因子,它是由HIF-1α和HIF-1β兩個亞基組成的異二聚體。HIF-1α在常氧時基本不表達,當機體處于缺氧狀態下則呈現高表達,促使細胞及組織發生一系列反應以使機體適應缺氧環境〔4〕。相關研究〔5,6〕顯示,在COPD患者肺組織中 HIF-1α mRNA及其蛋白質的含量均明顯升高,提示HIF-1α與缺氧性肺血管病變可能存在明顯相關性。HIF-1在缺氧早期即出現表達上調,其通過調控多種靶基因的表達,參與機體缺氧早期最先出現的分子水平適應性反應。諸多內源性細胞因子失衡都可以由缺氧誘發,如熱休克蛋白(HSP)90、內皮素(ET)-1、血管內皮生長因子(VEGF)生成增多,誘導型一氧化氮合成酶(iNOS)釋放異常等〔7〕,這一系列變化反應會引起肺血管收縮/舒張反應失衡,造成肺血管重塑,最終導致COPD肺血管病變的發生。本研究通過檢測COPD大鼠建模過程中肺組織HIF-1及其復合信號通路的基因表達情況,分析肺組織HIF-1復合通路基因表達改變情況與COPD肺血管病變發生發展的相關性。
1 材料與方法
1.1實驗動物 健康的雄性SD大鼠30只,體質量(200±20)g,購自維通利華生物科技股份有限公司,許可證號:SCXK(京)2016-0006。于溫度20~22 ℃,濕度50%左右環境中飼養。
1.2主要試劑及儀器 脂多糖(LPS,Sigma,批號:L2880);10% 甲醛固定液(索萊寶生物科技有限公司);無水乙醇(索萊寶生物科技有限公司);組織細胞總RNA提取試劑盒(天根生化科技有限公司);cDNA第一鏈合成試劑盒(天根生化科技有限公司);q-聚合酶鏈反應擴增試劑盒(天根生化科技有限公司);研究級倒置熒光顯微鏡(Olympus公司IX2-UCB型);超凈工作臺(ThermoFisher賽默飛世爾Herasafe KS型);高性能通用臺式冷凍離心機(ThermoFisher賽默飛世爾Sorvall ST 16R型);小型臺式離心機(ThermoFisher賽默飛世爾);-80 ℃超低溫冰箱(ThermoFisher賽默飛世爾ULT2186-6-V49型);熒光定時定量 PCR 儀(Bio-Rad伯樂JJQ5型)。
1.3分組及模型構建 將30只雄性SD大鼠隨機分為正常組10只和模型組20只,模型組后續分為造模6 w組和造模12 w組。正常組正常飼養,呼吸正常空氣;模型組每天在染毒箱內被動吸煙(大前門牌烤煙型過濾嘴香煙,卷煙條碼:6901028075916,煙氣煙堿量:0.8 mg) 10支,1 d分上下午煙熏2次,每次60 min,每煙熏6 d,休息1 d,共持續熏煙12 w。在實驗開始的第1天及第29天,使兩組吸入乙醚麻醉,在其氣道內分別緩慢滴注200 μl生理鹽水和200 μl LPS水溶液(1 mg/ml),且當日不再予被動吸煙,通過以上方法構建COPD大鼠模型。實驗過程中共有2只大鼠死亡,均來自模型組。
1.4標本采集 于造模前選取10只正常組大鼠,用2%戊巴比妥鈉 100 mg/kg腹腔麻醉后,酒精消毒,打開胸腔,收集肺組織,將右肺組織中葉于-80 ℃冰箱保存,以備后續檢測,左肺組織置于甲醛溶液中固定,至6 w隨機選取模型組大鼠10只進行上述操作,至12 w造模完成后,將剩余模型組大鼠進行上述操作。
1.5q-PCR檢測相關基因表達 各組肺組織進行q-PCR檢測,將肺組織在液氮中磨碎,參照組織細胞總RNA提取試劑盒說明書提取組織總RNA。以總RNA為模板,參照cDNA第一鏈合成試劑盒說明書合成cDNA第一鏈。使用熒光定時定量PCR儀以cDNA鏈為模板檢測相關基因表達。循環溫度為:94 ℃ 4 min、94 ℃ 30 s、56 ℃ 30 s、72 ℃ 30 s,循環數為40,以磷酸甘油醛脫氫酶(GADPH)為內參,采用2-ΔΔCt法比較分析各基因表達,PCR引物序列(3′-5′):GADPH上游引物:GGCAAGTTCAACGGCACAG,下游:CGCCAGTAGACTCCACGACA;HIF-1α上游引物:CGCAACTGCCACCACTGATGAA,下游:GTGAGGCTGTCCGACTGTGAGTA;HSP-90上游引物:TTTCCAACTCCTCAGACGCTC,下游:AGGGTT-CGGTCTTGCTTGTT;VEGF上游引物:CGACAAGGCAGACTATTCAACG,下游:GGCACGATTTAAG-AGGGGAAT;ET-1上游引物:TTCAGACTGGCAGAGGACCA,下游:TCACTTGCTACCAGCGGATG;iNOS上游引物:TCTGTGCTAATGCGGAAG,下游:TTGGTGTTGAAGGCGTAG。
1.6肺組織病理切片 經洗滌、脫水、透明、透蠟、包埋、切片、展片、貼片、脫蠟復水、染色、水洗、分化、漂洗、脫水、復染、脫水、透明、封藏等步驟制成病理切片,最終細胞核被蘇木素染成藍色,細胞質被伊紅染成粉紅色。
1.7統計學方法 采用SPSS21.0軟件行單因素方差分析,方差齊時用LSD法,方差不齊時用Dunnett法。
2 結 果
2.1COPD模型造模后不同時間點肺組織病理學變化 HE染色結果顯示,正常組肺組織中支氣管及肺泡結構完整,黏膜下未出現明顯的炎細胞浸潤,而在造模6 w后,相較正常組,支氣管結構仍較為完整,但在黏膜下出現了明顯的炎細胞浸潤(黑色箭頭),與此同時,肺泡出現結構不一,肺泡壁變薄或斷裂引起部分肺泡擴大融合形成肺大泡(紅色箭頭),肺血管表現出不同程度增生,管壁增厚或管腔變形,管壁及周圍可見較多炎性細胞浸潤,微血管數量明顯增多(黃色箭頭);造模12 w后,COPD模型支氣管結構出現一定程度損傷,部分支氣管上皮細胞脫落(藍色箭頭),黏膜下出現大量炎細胞浸潤(黑色箭頭),肺泡結構紊亂,并進一步擴張融合形成大量肺大泡(紅色箭頭),且肺血管持續增生,其不論是管壁增厚還是管腔變形均較前有所加重,微血管數量進一步增多(黃色箭頭)。見圖1。
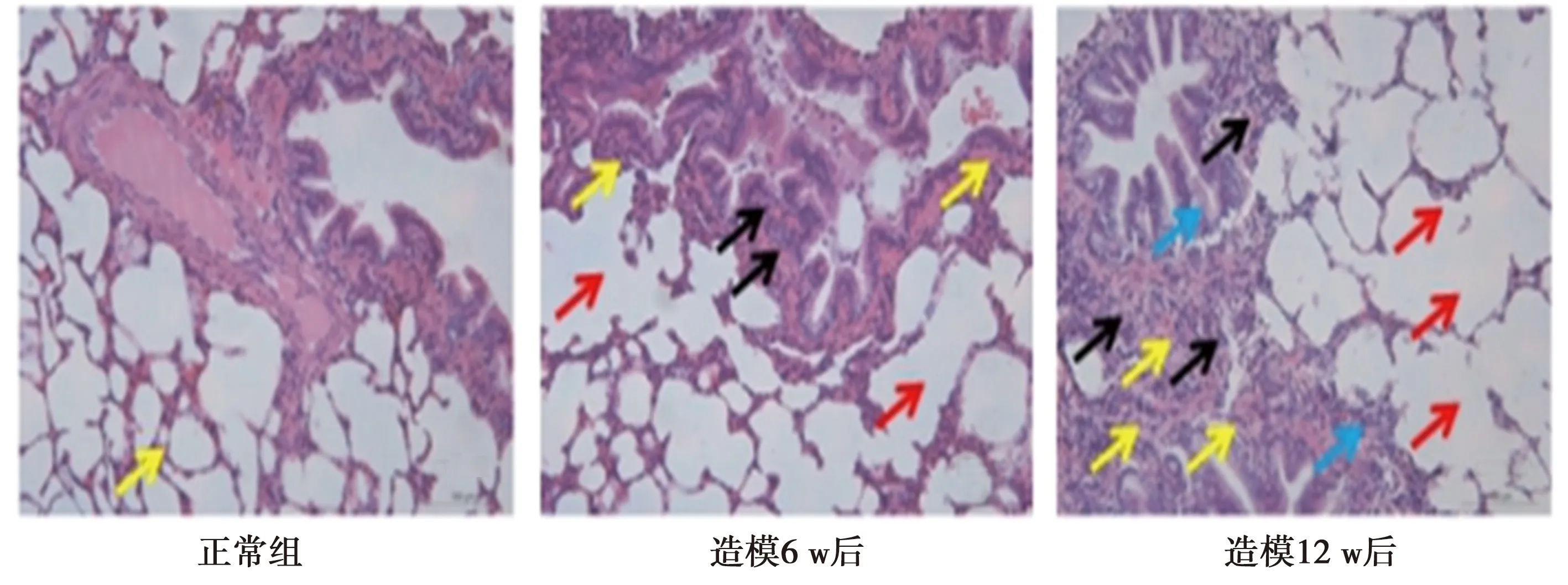
圖1 COPD模型造模6、12 w時肺組織(HE染色,×200)
2.2造模后不同時間點肺組織基因表達水平 與正常組相比,造模6、12 w后肺組織中HIF-1α、HSP90、VEGF、ET-1 mRNA表達顯著上調(P<0.05,P<0.01),且造模12 w后肺組織中HIF-1α、VEGF、iNOS mRNA表達與造模6 w時相比顯著上調(P<0.05)。與造模6 w時相比,造模12 w后肺組織中HSP90、ET-1 mRNA表達無顯著差異(P>0.05)。與正常組相比,iNOS mRNA表達在造模6 w時無顯著差異(P>0.05),造模12 w后肺組織中iNOS mRNA表達顯著上調(P<0.05)。見表1。
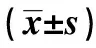
表1 造模后不同時間點肺組織中HIF1-α、HSP90、VEGF、ET-1及iNOS mRNA表達水平
2.3COPD模型造模后不同時間點肺組織中HIF-1復合通路相關基因表達水平 分別收集正常組、造模6、12 w組肺組織,q-PCR法檢測COPD模型造模后不同時間點HIF-1復合通路關鍵基因HIF-1α、HSP90、VEGF、iNOS及ET-1 mRNA表達,GAPDH被選取作為內參。各基因擴增曲線見圖2。可見各基因引物擴增曲線重復性良好,溶解曲線呈單峰,可用于后續實驗檢測。
3 討 論
HIF-1是廣泛分布于人體內的一種異源二聚體轉錄因子,由120 kU的HIF-1α 和91~94 kU的HIF-1β兩個亞單位組成〔8〕。HIF-1蛋白只有機體處于缺氧狀態下才具有活性,常氧下HIF-1蛋白很快失去活性。HIF-1的調控活性取決于它的α亞基,激活的HIF-1α通過增加與下游基因缺氧反應元素的結合,對多種轉錄因子的表達和翻譯起到調控作用〔9〕。研究發現〔10〕隨著COPD病程進展,血液及肺組織中HIF-1α的表達均進一步升高,平均肺動脈壓也隨之增加,體現血管重構的指標血管壁厚度/血管外徑(WT)%及血管壁橫斷面積/血管橫斷面積(WA)%也明顯升高,提示HIF-1α與肺血管病變存在顯著的相關性。與本研究結果一致,進一步證明HIF-1復合信號通路參與了COPD肺血管病變的發生發展。
當機體輕度缺氧時,HIF-1α的表達即開始上調,伴隨著機體持續缺氧的加重,當機體細胞質內含氧量低于5%處于重度缺氧狀態時,HSP90表達開始上調。HSP90作為HIF-1α的增強子,對HIF-1α也同樣起著調節作用,其可以與HIF-1α的PAS區特異結合,使HIF-1α結構更加穩定,從而干預HIF-1α的泛素化降解〔11〕。故如果低氧持續得不到改善,將導致HIF-1α生成增多,而降解減少,形成惡性循環。HSP90活性被破壞可導致蛋白酶體降解HIF-1α,研究發現〔12〕HSP90的抑制劑17-AAG和17-DMAG能夠促進HIF-1α蛋白的降解,進而抑制血管的生成。作為HIF-1的下游基因,現已有研究證實,HSP90同樣具有促進血管生成的作用,是血管生成的關鍵分子之一〔13〕。在HSP90低氧時的增強作用下,HIF-1α結構更加穩定,HIF-1復合通路在低氧時得以進一步過量表達。
有研究發現〔14〕,在合并肺動脈高壓的COPD患者中,呼出氣冷凝液及循環血液中的ET-1水平均較常人明顯上升,并且其濃度與肺動脈的收縮壓或平均壓表現出明顯的關聯性。ET-1作為一種由21個氨基酸構成的生物活性多肽,是至今所發現的功效最強的縮血管生物因子,廣泛分布于肺血管內皮細胞中〔15〕。低氧狀態時,HIF-1α能夠與ET-1啟動子區域的 HIF-1結合位點特異性結合,激活內皮細胞ET-1表達〔16〕。ET-1的過量表達會引起肺動脈強烈收縮,促進肺動脈平滑肌細胞、血管外膜成纖維母細胞及膠原纖維的增殖,并可通過激活絲氨酸/蘇氨酸蛋白激酶(Rho)減少血管和肺泡的生長,進而引起肺血管病變〔17〕。
相關研究顯示〔18〕,VEGF在COPD患者血及痰液中的含量明顯高于健康人群,且其升高水平與肺動脈高壓的發生密切相關,提示VEGF參與肺血管病變的發展。VEGF的增強子中含有可與HIF-1α結合的低氧反應元件,當與增強子結合后可增強其蛋白水平的表達及轉錄,此外,HIF-1可在缺氧情況下有效增強VEGF mRNA穩定性,促使其缺氧時大量表達。VEGF是一種具有多種功能效應的細胞因子,其主要在人體血管豐富的器官及組織中分布,可以介導血管生成與血管重塑的發生〔19〕。有研究表明〔20〕,VEGF能夠通過上調ET-1的表達,增強血管基質金屬蛋白酶活性,以促進平滑肌細胞的增殖,參與血管重塑。作為已發現的功效最強的血管通透劑,它不但可以刺激血管內皮細胞的增殖和遷移,還能誘導體內血管形成,在肺血管病變過程中扮演著重要角色〔21〕。
機體正常狀態下僅合成少量的NO,其作為重要的細胞信使,少量內源性NO能夠舒張血管,改善組織微循環,發揮調節肺血流及免疫防御的功效。而當機體處于缺氧狀態時,HIF-1α的大量表達可以激活NOS的轉錄,使其表達量顯著增加,釋放大量NO,產生自由基,損傷內皮細胞,造成血管活性物質的異常改變及血管收縮-舒張功能失調,使肺血管發生痙攣,引起肺動脈壓升高〔22〕。NO也是一種致炎因子,在炎癥反應中,NO可以激活白細胞并促進其遷移,高濃度NO的毒性作用會增加血管內壁的通透性,誘發血管炎癥,致使血管擴張、內膜增厚,最終引發肺泡-肺毛細血管功能的異常改變〔23,24〕。
綜上,COPD肺血管病變的發生、發展與HIF-1復合信號通路的表達相關,且隨著COPD肺血管病變時間的延長,HIF-1復合信號通路的表達也逐漸升高,說明HIF-1及其下游靶基因在缺氧性肺血管病變形成中起重要作用,對研究相關疾病的發病機制有重要意義,可將HIF-1作為靶點,通過抑制其復合通路的表達,以改善或阻止肺血管的重構,從而起到治療肺心病等肺血管疾病的作用。而HIF-1復合通路復雜,下游靶基因眾多,是否還存在其他下游基因參與介導了缺氧性肺血管病變過程仍值得深入研究。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
