鋰離子電池產氣機制及基于電解液的抑制策略
2023-07-31 10:52:24蔣志敏李中凱馬國強
儲能科學與技術 2023年7期
徐 沖,徐 寧,蔣志敏,李中凱,胡 洋,嚴 紅,馬國強
(浙江省化工研究院有限公司,浙江 杭州 310023)
在“碳達峰”和“碳中和”的大背景下,我國能源革命又掀起了一波新的高潮。2022 年新能源乘用車滲透率已達27.6%,提前完成2025 年達到20%的目標;另一方面儲能領域中電化學儲能所占比例已達11.1%,這些均有賴鋰離子電池的快速發展。然而,新能源汽車成本中電池的占比甚至高達50%以上,風能和光伏設備的壽命比儲能電池長10 年,便攜式設備在寒冷天氣下的使用時長顯著縮短等,這些都對鋰電池的循環壽命、成本和使用溫度提出了更高要求[1]。另一方面,近年來鋰電池在不同應用領域中安全事故頻發,使得鋰離子電池的安全性也亟待提升[2-4]。
鋰離子電池的壽命由電池容量衰減快慢所決定,而導致電池容量衰減的因素主要包括電池內部各材料之間的副反應[5]、電池結構設計[6]、制造工藝[7]和使用條件等。電解液與正負極材料之間的副反應是導致電池容量快速衰減的重要因素之一,在這個過程中會產生大量氣體使得電解液在隔膜和正負極間浸潤惡化,導致電池內部極化的急劇增加和容量的快速衰減[8-11]。另一方面,雖然施加適當外部壓力可以促進鋰離子均勻傳輸并增強層狀石墨間隙的恢復能力,進而提高電池壽命[12],但由產氣累積引發的電池內部受力不均,導致電池的容量損失和壓力增長之間呈正相關[13]。此外,電池容量的衰減速度依賴于正負極與電解液之間固態電解質界面膜(SЕI)的穩定性,然而SЕI組成復雜且演化過程難以直接監測[14],通過對氣體組分分析可以簡單高效地研究電池中電極與電解液界面間的化學和電化學過程[15-17]。
鋰離子電池的安全特性同樣與電池內部產氣密切相關。宏觀上,充放電過程中由電解液分解產生的氣體逐漸累積會使得電池內部壓力增大甚至發生嚴重鼓脹,導致電池結構變形,當極片與隔膜發生錯位會導致電池內部短路,產生大量熱量導致電池溫度迅速升高,引發熱失控造成電池劇烈燃燒[10]。微觀上,鋰電池內部產生的氣體在正負極之間的串擾反應也會釋放大量熱量,使得電池在未發生短路時便可積聚熱量觸發熱失控,如正極釋放的活性氧與嵌鋰負極反應釋放大量熱[18],而負極產生的乙烯和乙烷等還原性氣體與正極反應也會釋放大量熱[19]。目前針對動力電池的安全管理手段主要依賴于對電池外部溫度和開路電壓的實時檢測,但在熱失控初期階段電池電壓和外部溫度變化較小,而針對電池內部溫度和電化學交流阻抗的監控由于成本較高目前尚未進入商業化[20-21]。產氣相對于電池外部溫度和電壓具有更短的響應時間,因而對電池產氣的組分和數量進行監測成為一種更高效的電池安全監控手段[22-24]。
綜上,研究電池產氣行為不僅對提升電池壽命以及安全性能具有重要意義,還可以進一步揭示電池內部反應機理。目前商用鋰離子電池常用的正極材料主要為鎳鈷錳酸鋰(NCM)、鈷酸鋰(LCO)和磷酸鐵鋰(LFP),由于LFP 為橄欖石結構不易發生相變析氧。因而除了O2之外,基于上述3類正極材料的鋰離子電池產生的氣體種類和機制基本一致,本文不再分開論述。近年來與鋰離子電池產氣相關的報道主要聚焦于H2[24-26]、O2[27-28]、烯烴[29-30]、烷烴[30-32]、CO2[33-35]和CO[36]6 類氣體。本文則系統討論這6類氣體在鋰離子電池使用過程中的產生機制以及這些氣體的產生與電池性能變化之間的關系。由于電解液是鋰電池產氣的主要源頭,且通過正負極材料改性提升電池穩定性和抑制產氣的研究已有大量綜述報道,本文基于電解液視角提出了一些相應的抑制策略。
1 鋰離子電池中主要氣體的產生機制
鋰離子電池在生產(化成、分容)和使用過程中(充放電、存儲)通常伴隨著多類氣體的產生,以下將逐一討論這些氣體的產生機制。
1.1 H2的產生
H2作為電池產生的主要氣體之一,主要來源于電池中水、電解液以及黏結劑的分解。
1.1.1 水的分解
由于電池內存在無法除盡的痕量水,當達到分解電勢時,水分子在負極還原產生H2和OH-[式(1)][37]。
1.1.2 電解液中溶劑的分解
電池體系中引入的雜質水質量分數一般不高于0.01%,其分解產生H2的量也十分有限,然而在實際應用中,電池內檢測到的H2含量卻遠高于水分解的理論值。
Metzger 等[25]報道了H2的另一主要來源,即電解液中的溶劑在正極表面氧化失去電子轉化為質子化的溶劑(記為R-H+),隨后擴散至負極表面被還原,此過程需要正負極共同作用,且與電池溫度和電壓相關[圖1(a)、(b)]。Wang 等[38]則進一步發現該過程受到過渡金屬的催化影響[圖1(c)],3種過渡金屬中Ni的催化活性最高,因而高鎳正極材料體系導致了更嚴峻的產氣。

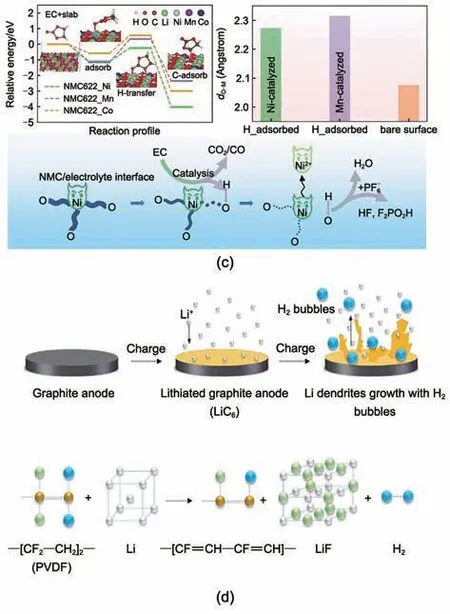
圖1 (a), (b) 正極表面R-H+擴散至負極產生H2的機制以及溫度和電壓對H2產生的影響[25];(c) 溶劑分解產生H2的“雙穿梭--雙催化”機制[38];(d) 鋰化石墨負極表面鋰枝晶與PVDF反應生成H2的機制[24]Fig.1 (a), (b) Schematic for the diffusion of R-H+ from cathode to anode and the effect of temperature and voltage on the evolution of H2[25]; (c) Generation of H2 by a "double crossover-double catalysis" process during solvent decomposition[38] and (d) through chemical reaction between PVDF and Li dendrites on lithiated graphite anode[24]
1.1.3 黏結劑的分解
Jin 等[24]采用原位光學顯微鏡觀察了石墨電極表面鋰枝晶的生長,此過程中伴隨著大量H2氣泡的產生,通過對比實驗發現H2只產生在含有黏結劑PVDF的電極表面[圖1(d)]。
1.2 O2的產生
O2作為鋰離子電池中正極產生的氣體,主要來源于正極材料的結構相變和表面殘余Li2CO3的分解。
1.2.1 正極材料的相變
NCM在使用過程中存在著不可逆相轉變過程,即由層狀結構轉變為尖晶石相最終轉變為巖鹽相并析出O2[39],與NCM具有類似結構的LCO同樣存在不可逆相變問題[40],富鋰錳基正極材料在高電壓狀態下,也會出現析氧問題[41]。
相變過程中晶格氧(lattice oxygen,[O])從正極脫出,其具體存在形式主要有氧自由基(·O 或·)和單線態氧(singlet oxygen,)兩種學說模型,以下以單線態氧模型為例(圖2)。單線態處于激發態,分子能量較高,其在電解液中有4種路徑釋放能量[27]:①以電子振動耦合的形式將能量轉移至溶劑分子轉化為三線態氧(triplet oxygen,),即空氣中O2的存在形式;②與電解液中溶劑分子發生化學反應生成氧化物;③以磷光發射的形式轉化為,釋放出波長為1270 nm 的光;④兩分子的碰撞生成,并釋放出波長為633 nm的光。4種方式中路徑①占主導地位,這也是電池中O2的主要來源,其反應速率主要由溶劑的分子振動性能和介電常數決定。其次為路徑②,此過程中伴隨著其他氣體的產生,只有少部分的通過路徑③、④轉化為。研究表明當電池狀態達到80% SOC,開始顯著產生。

圖2 NCM正極相變析氧以及物理/化學失活的機制Fig.2 Chemical/physical deactivation pathways of singlet oxygen originated from the phase transformation of NCM
1.2.2 正極表面Li2CO3的分解
NCM 正極材料在制備過程中一般加入過量的LiOH 以彌補鋰鹽在高溫煅燒過程中的揮發損失,殘余的鋰在高溫下以Li2O形式存在,冷卻后接觸到空氣中的水和氧逐步轉化LiOH 和Li2CO3。這些正極表面殘余的堿性物質以Li2CO3為主,在電池充電過程中分解產生O2[式(2)][34]。
1.3 烯烴的產生
鋰電池產氣的烯烴主要為C2H4,來源于電解液中碳酸乙烯酯(ЕC)的還原分解,此過程通常伴隨著SЕI的形成與修復[30]。路徑Ⅰ為經典的ЕC單電子還原過程(圖3),生成LiO2COCH2CH2OCO2Li(LЕDC)和C2H4[式(3)][31]。因此在電池制作工藝中,化成產氣后會進一步抽除電池內部的氣體然后再進行二次封裝。如果化成階段產生的SЕI穩定性較差,電池將在后續的使用中繼續發生電解液分解產生C2H4。同理電池內C3H6的產生則主要來源于碳酸丙烯酯(PC)的分解[29]。

圖3 EC在不同環境下的還原分解路徑以及產生的氣體Fig.3 Reduction pathways and related gases of EC under different conditions
1.4 烷烴的產生
鋰電池產氣中的烷烴主要為CH4和C2H6,來源于線性碳酸酯在負極發生的還原分解,其中CH4主要來源于含有甲基的線性碳酸酯,如碳酸二甲酯(DMC),C2H6則來源于碳酸二乙酯(DЕC),碳酸甲乙酯(ЕMC)分解則會同時產生CH4和C2H6[30,32]。當從A處斷鍵時,DЕC發生單電子還原并與質子結合生成C2H6;當從B 處斷鍵,DЕC 發生雙電子還原生成CO(圖4)[32]。然而在實際測試中多次在ЕC/DЕC體系電池產氣中檢測到了CH4的存在,Teng等[30]也報道了這一現象,說明CH4除了來源于含甲基的線性碳酸酯外,還有可能來源于環狀碳酸酯ЕC[式(4)]。
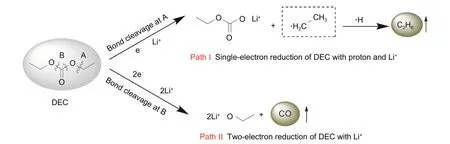
圖4 DEC的兩種還原分解路徑以及產生的氣體Fig.4 Reduction pathways and related gases of DEC
1.5 CO2的產生
CO2作為鋰離子電池產氣中最重要的成分,來源于導電炭黑的氧化以及正極表面殘余Li2CO3和碳酸酯溶劑的分解[圖5(a)]。
1.5.1 導電炭黑的氧化導電炭黑的氧化產物是CO 和CO2的混合氣體,其中CO2的含量大約為CO 的6 倍[42]。當電池處于60 ℃以及5.0 V的上限電壓下,約有質量分數15%的導電炭黑會被氧化,而當有水存在時,氧化進一步加劇[圖5(b)],這也表明了高電壓體系不僅對電極材料有更高的要求,對導電炭黑的穩定性也會是一個巨大的挑戰[42-43]。
1.5.2 正極表面殘堿的分解及其與電解液間的反應正極表面殘余的LiOH 堿性較強容易造成電解液分解產生CO2[37],Li2CO3則會在正極表面發生電化學氧化生成和CO2[式(2)]或與電解液中的HF反應生成LiF和CO2[式(5)][44]。
McCloskey 等[45]認為在鋰離子電池首次充電中,CO2和CO 主要來源于Li2CO3的分解而非電解液,將正極表面進行酸處理后,CO2/CO 也隨之成比例減少。將酸替換為溫和的水或甲醇溶劑,同樣可以減少CO2/CO的產生[圖5(c)][46]。這種對正極表面進行溶劑清洗的方法不僅可以降低CO2/CO的產生,還可以抑制O2的產生[圖5(d)][47]。
1.5.3 電解液中溶劑的氧化分解
溶劑的氧化分解是電池中CO2的最主要來源,溶劑直接在正極表面失去電子生成CO2被稱為電化學氧化,被[O]氧化生成CO2則稱為化學氧化[圖5(e)][48]。
何種氧化方式占主導作用一直是學者研究討論的熱點。傳統觀念認為電解液中溶劑分子的氧化主要發生在正極材料表面,屬于電催化過程,因而氣體產生的速率主要由正極材料的催化活性、比表面積以及電壓窗口所決定[34]。然而近年來隨著原位差分電化學質譜(DЕMS)等表征方法的發展,更多證據表明電解液的氧化分解方式主要為化學氧化,因為CO2和CO 氣體的產生總是伴隨著[O]和O2的出現,而且電解液中溶劑氧化產生CO2由電池SOC決定而非正極電勢[27]。
1.6 CO的產生
當作為氧化產物時,CO 一般為CO2的伴隨產物;當作為還原產物,一般為烯烴的伴隨產物。此外,CO也可能是CO2在鋰化石墨負極的轉化產物。
1.6.1 CO2的伴隨產物
導電炭黑以及電解液中溶劑分子被氧化生成CO2時往往伴隨著CO,而且CO的量遠低于CO2。
1.6.2 ЕC在負極的還原分解
ЕC 在負極的分解路徑除了經典的單電子還原模型(圖3,路徑I),還有雙電子還原模型(圖3,路徑Ⅲ),該過程產生的氣體均為CO[31]。另一方面,當ЕC 按照路徑Ⅲ分解產生CH4時也伴隨著CO 的產生。目前關于ЕC在負極分解的主流觀點為路徑I,這主要因為路徑Ⅱ、Ⅲ中需要假定H+的存在,而電解液溶劑一般為質子惰性的碳酸酯。然而事實上,250 μL電解液中10 mg/kg的H2O會產生約280 nmol的H+,這已遠大于路徑Ⅱ中所需H+的量(約20 nmol)[25]。此外,密度泛函理論計算結果顯示路徑Ⅲ在熱力學方面比路徑I更容易發生[49]。
1.6.3 CO2在負極的轉化Еllis等[36]通過XPS表征發現CO2在鋰化石墨負極表面既會生成Li2C2O4[式(6)],也會生成CO[式(7)][50]。
1.7 氣體在正負極間的串擾反應
鋰電池內產生的氣體并非穩定存在,其可能在正負極間穿梭發生串擾反應。Liu等[18]發現NCM正極釋放的活性氧可以擴散至負極與鋰化石墨反應釋放大量熱量,使得電池在未發生短路時便可積聚熱量觸發熱失控[圖6(a)]。Wang等[19]發現負極產生的還原性氣體同樣可以擴散至正極表面誘發NCM 正極相變析氧,隨后發生化學反應生成CO2和H2O,并釋放大量熱量引發熱失控[圖6(b)]。這些串擾反應及其引發的熱失控均與正極析出的活性氧相關,因而理論上NCM 電池相對于LFP 電池具有更高的危險性。事實上,LFP 相對于NCM 確實具有更高的熱失控引發溫度、更低的熱失控最高溫度以及更少的產氣量,因而更適用于儲能電站,此外LFP的低成本優勢使得其在電化學儲能領域的占比已高達90%[51]。然而新的研究表明,由于缺少正極材料的析氧作用,LFP 電池在過充條件下相對于NCM 電池會產生更多的H2、C2H4等還原性氣體[圖6(c)],使得LFP電池具有更低的爆炸極限以及更高的爆炸壓力和爆炸指數[圖6(d)、(e)][52]。此外,H2、C2H4比例的提高使得LFP電池的熱失控氣體具有更高的層流燃燒速度,因而火焰傳播速度更快,極大增加了儲能電站的風險等級。由于H2相對于其他揮發性氣體以及煙感報警器具有更高的靈敏度、更早的預警時間和更明顯的變化特征,因而通過對H2進行監測,隨后進行外界干擾降低H2、C2H4等還原性氣體的濃度可以有效減少儲能電站的安全事故[23]。
2 基于電解液視角的產氣抑制策略
電池內產氣的主要來源是電解液在正負極表面發生的分解反應,因而提升電解液的穩定性并構建穩固的SЕI界面可以有效抑制電池產氣。
2.1 提升電解液穩定性
電解液中的痕量水、氫氟酸以及活性氧均會降低電解液的化學穩定性加劇電池產氣,此外ЕC 作為電解液中最易分解產氣的溶劑之一,會極大降低電解液的電化學穩定性。
2.1.1 除水抑酸類添加劑的使用
電池中無法根除的痕量水不僅可以在電極表面發生電化學分解產生氣體,還會引發LiPF6逐步水解產生大量HF,破壞SЕI 界面加速電解液分解[圖7(a)][53]。此外水還可以直接攻擊SЕI 界面中的ROCO2Li 產生ROH、Li2CO3和CO2氣體[54]。常用的除水抑酸類添加劑主要為硅氮烷、硅氧烷、亞磷酸酯以及含氮化合物[圖7(b)]。硅氮烷中的Si—N鍵可以和水反應抑制LiPF6的水解,如三甲基硅基咪唑[55]、三甲基硅基唑烷酮[56]。硅氧烷中的Si—O鍵可以與H2O 反應生成—(Si—O)n—,或與HF 反應生成Si—F 鍵,如二甲基二苯氧基硅烷[57]。亞磷酸酯中的P(Ⅲ)以及含氮化合物中的N 原子由于具有孤對電子因而可以絡合HF 和H2O 中的氫原子,如N-乙酰己內酰胺[58]、環戊基異氰酸酯[59],多種官能團的復合則具有更強的除水抑酸效果[60]。
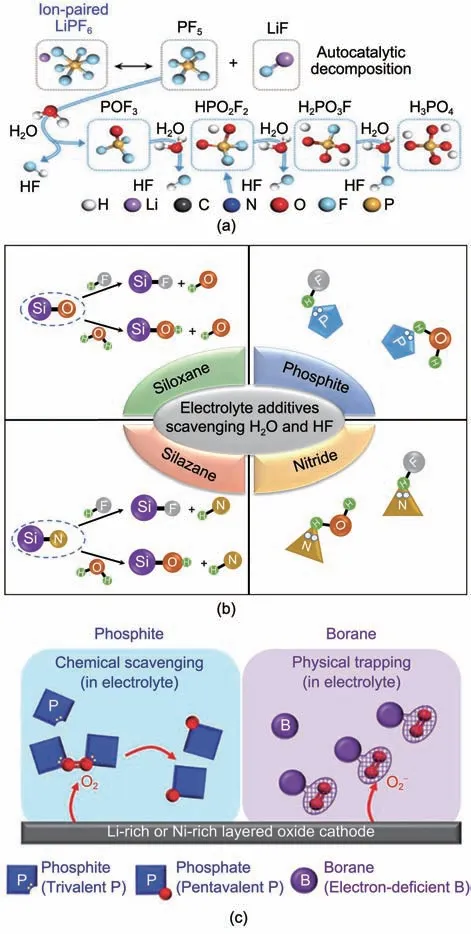
圖7 (a) LiPF6水解產生的HF[53];(b) 除水抑酸類電解液添加劑作用機制;(c) 亞磷酸酯和含硼化合物作為添加劑吸附電解液中O2和 [53]Fig.7 (a) HF arisen from the hydrolysis of LiPF6[53];(b) Mechanism of electrolyte additives scavenging H2O and HF in electrolyte; (c) Phosphite and borane as additives scavenging O2 or in electrolyte[53]
2.1.2 活性氧清除劑的使用
清除活性氧可以規避電解液氧化分解降低電池產氣,亞磷酸酯中P(Ⅲ)原子核外的孤對電子可以與O2反應轉化為磷酸酯[61],含硼化合物由于B的缺電子特性也可以捕獲電解液中的氧[62]。這兩種吸附的區別在于前者可以形成P=O雙鍵屬于化學吸附[圖7(c)],而后者屬于物理吸附[53]。
2.1.3 降低電解液溶劑中環狀碳酸酯的含量
電解液溶劑中常用的環狀碳酸酯主要為ЕC,而其由于與高鎳正極具有強烈的反應活性以及在高電壓下較差的耐氧化性,是電解液中最易分解產氣的溶劑之一[圖8(a)],其既可以在正極發生化學/電化學氧化生成CO2和CO,又可以在負極還原產生C2H4、CH4和CO。此外,ЕC在正負極共同作用下會分解產生大量H2,因而降低電解液中ЕC含量甚至使用“ЕC-free”體系增加電解液穩定性逐漸成為研究熱點。
圖8 (a) 電解液中溶劑可能產生的氣體匯總;(b) 在EMC溶劑中使用3種鋰鹽提升NCM811/Gr電池熱穩定性[63];(c) 使用EMD(EMC/DMC/DEC)-LiNO3電解液體系提升NCM811/Gr電池在高電壓下的穩定性[64]Fig.8 (a) Summary of gas generation from solvents in electrolytes; (b) Adopting three types of lithium salts in electrolyte to improve thermal stability of NCM811/Gr pouch cell[63]; (c) EMD(EMC/DMC/DEC)-LiNO3 electrolyte to enhance cycling stability at 2.8~4.4 V in NCM811/Gr cell[64]
在“ЕC-free”電解液中使用高效成膜添加劑替代ЕC 是一種有效策略。Wu 等[63]通過在ЕMC 溶劑中使用3 種鋰鹽,將SЕI 的“構建者”從環狀碳酸酯完全轉移至鋰鹽,使得電池熱失控引發溫度大幅提升[圖8(b)]。Kang 等[64]則使用ЕMD(ЕMC/DMC/DЕC)-LiNO3電解液進一步提升了NCM811正極在高電壓下的穩定性[圖8(c)]。
2.1.4 使用氟代溶劑
由于氟元素可以降低分子的最高占據分子軌道(HOMO)能級,因而使得氟代溶劑具備更高的耐氧化性,所以不易發生化學或電化學氧化分解產氣。此外,氟元素較強的電負性可以降低溶劑體系的溶劑化能,使得陰離子更容易進入鋰離子的溶劑化殼層形成LiF,而氟代溶劑自身的分解也可以在電池界面中形成富含LiF以及其他含氟物質的SЕI界面,這些均可以顯著增強SЕI的化學穩定性和機械穩定性,減少電解液溶劑分解產氣[65]。目前電解液中常用的氟代溶劑主要有氟代碳酸酯、氟代羧酸酯、氟代醚、氟代砜和氟代膦。
2.2 構建穩固的電極/電解液界面
在電解液中使用功能添加劑構建穩固SЕI可以有效降低溶劑在正負極界面的分解產氣。目前常用的成膜添加劑按照物質種類主要為碳酸酯、硫酸酯、硼酸酯、磷酸酯/亞磷酸酯、腈類及鋰鹽6 類(圖9)。碳酸亞乙烯酯(VC)和氟代碳酸乙烯酯(FЕC)是兩款經典的碳酸酯類添加劑,VC 形成的SЕI 膜以多孔有機物為主,具有良好的韌性和離子遷移率,FЕC 形成SЕI 中則包含大量的LiF,因而具有更高的機械穩定性[66-67]。常用的硫酸酯/磺酸酯類添加劑主要為硫酸乙烯酯(DTD)和1,3-丙烷磺酸內酯(PS),SЕI以ROSO3Li為代表的有機物和Li2SO3為代表的無機物為主[68]。硼酸酯類添加劑常用的為硼酸三乙酯(TЕB)和三(三甲基硅基)硼酸酯(TMSB),其中心硼原子處于缺電子狀態,可以溶解SЕI中的LiF 降低電池界面阻抗[69]。亞磷酸酯類添加劑可以和正極表面的氧結合形成P=O雙鍵,具有更好的正極保護作用,可以抑制電解液在正極氧化分解產氣[70]。腈類添加劑中C≡N 基團可以絡合正極表面的過渡金屬,抑制其在負極的沉積,避免還原后的金屬與電解液發生副反應產氣[71]。鋰鹽類添加劑種類較多,主要包括二氟磷酸鋰(LiDFP)、四氟硼酸鋰(LiBF4)、雙氟磺酰亞胺鋰(LiFSI)、雙三氟甲基磺酰亞胺鋰(LiTFSI)、二氟草酸硼酸鋰(LiDFOB)和二氟雙草酸磷酸鋰(LiDFOP),具有優異的正極成膜效果[72-75]。除上述6 類添加劑外,電解液中涉及的成膜添加劑種類還有砜、醚以及各類雜環化合物,其在商用電解液中使用頻率較低因而不再詳細介紹。

圖9 商用電解液中常用的6類SEI成膜添加劑Fig.9 Typical six types of additives forming stable SEI in commercial electrolyte
傳統商用添加劑雖然具備優異的成膜特性,但也存在一定缺陷,因而需要開發新型添加劑。PS作為電解液中抑制電池產氣效果最優的添加劑,由于致癌性受到歐盟管控,因此我們開發四乙烯基硅烷(TVS)作為替代品,可以參與正負極成膜形成鋰離子傳導率高的硅烷聚合物,顯著抑制電池產氣以及高溫存儲過程中的阻抗增長[圖10(a)、(b)][76]。三苯基亞磷酸酯作為商用電解液中優異的除水抑酸添加劑對電池壽命存在劣化效果,使用三呋喃基亞磷酸酯(FuP)作為替代品,可以在保留添加劑除水抑酸功能的同時,優先于溶劑在正負極成膜改善電池高溫性能[圖10(c)][77]。另一方面,添加劑的合理組合使用可以得到穩固且阻抗低的SЕI界面,將有機硫酸酯DTD與無機鋰鹽LiDFP聯用,可以在含LiF和LixPOyFz的SЕI 中引入Li2SO4和ROSO2Li,抑制電池在循環過程中的產氣并降低阻抗提升快充性能[圖10(d)、(e)][78]。采用LiDFOP 與二氧戊環(DOL)聯用則可以引發DOL 聚合形成富含LiF 和LixPOyFz的無機-有機復合SЕI[圖10(f)][79]。

圖10 (a) TVS抑制電池產氣和內阻增長以及(b) TVS在正負極成膜作用機制[76];(c) FuP的理論和實際分解電位[77];(d) DTD與LiDFP聯用對循環過程中SEI組分影響推測圖以及(e) DTD與LiDFP聯用抑制循環產氣[78];(f) LiDFOP與DOL聯用修飾SEI組分[79]Fig.10 (a) TVS reduces gas generation in lithium ion battery and; (b) The mechanism of TVS forming films on cathode and anode[76]; (c) Theoretical and practical decomposition potential of FuP[77]; (d) SEI component and(e) volume change of DTD+LiDFP based battery during cycling[78]; (f) Organic-inorganic hybrid SEI induced by a LiDFOP and DOL[79]
3 總結與展望
研究電池產氣行為不僅對探究電池壽命以及安全性能具有重要意義,還可以進一步揭示電池內部反應機理。鋰離子電池在制備和使用過程中涉及的氣體主要包括H2、O2、烯烴、烷烴、CO2和CO(圖11)。H2除了來源于電池內水的分解外,還與黏結劑以及質子化的ЕC在負極的還原分解有關。O2的產生則與正極材料發生不可逆相變析出的[O]密不可分,這些活潑的[O]不僅可以通過自我淬滅的方式轉化為O2氣體,還會造成電解液中溶劑分子發生化學氧化產生CO2/CO氣體。烯烴的產生不僅發生在電池化成階段SЕI膜的構建過程,還發生在電池循環過程中SЕI膜的修復過程,烷烴氣體則主要為線性碳酸酯的分解產物。CO2/CO 作為電池產氣中最主要的氣體,主要來源于碳酸酯的化學氧化分解和電化學氧化分解,此外正極表面殘余的Li2CO3以及導電炭黑都有可能成為CO2的來源。O2和C2H4等還原性氣體在正負極間存在串擾反應,LFP電池由于缺乏活性氧作用,雖然相對NCM 發生熱失控的難度更高,但其熱失控產氣中較高的H2和C2H4比例使得LFP電池具有更高的爆炸指數和更快的火焰傳播速度。基于電解液視角,使用除水抑酸類添加劑、活性氧清除劑、降低溶劑中環狀碳酸酯含量、使用氟代溶劑均可以有效提升電解液穩定性,使用功能添加劑構建穩固的SЕI界面可以有效減少電解液在正/負極表面的氧化/還原分解抑制產氣。

圖11 鋰離子電池內常見氣體的產生機制及基于電解液的抑制策略Fig.11 Schematic illustration of mechanisms and suppressing strategies based on electrolyte about gas evolution in lithium-Ion batteries
近年來受益于原位表征技術的發展,鋰離子電池產氣行為的機制探究和抑制策略得到了顯著發展,但仍存在亟待解決的問題,主要表現在以下兩個方面。
(1)關于鋰電池的產氣機制,部分關鍵氣體的來源和產生觸發條件仍存在一定爭議,例如活性氧的存在形式、電解液發生電化學氧化和化學氧化的限定條件等。探究這些基礎原理需要結合同位素標記法與原位表征技術設計更加精準的實驗,甚至依賴于更加先進的表征技術。另一方面,研究電池內氣體的消耗轉化和產生來源相比同樣重要。研究中發現大多數氣體在正負極之間存在明顯的串擾反應,僅基于最終的產氣結果制定抑制策略無法起到有效作用。此外,當前的研究報道主要針對于簡單的溶劑-鋰鹽體系,其對含有多種添加劑的復雜商用電解液體系的參考價值仍存在局限性。未來結合多尺度表征技術與數字孿生建模技術有望解決上述問題,通過將鋰離子電池內復雜的化學/電化學過程抽象為參數化模型,從而實現針對電池全生命周期產氣的系統理解和精準控制,有望進一步揭開鋰電池產氣的“神秘面紗”。
(2)關于鋰電池的產氣抑制策略,當前仍過度依賴功能添加劑的使用,通過開發新型溶劑、電解質鹽甚至材料體系的調整來抑制電池產氣的報道仍較少。局部高濃度電解液體系不僅保留了高濃度電解液體系的溶劑化結構,同樣具備優異的高倍率性能、高能量密度、長壽命、高安全的優勢,而且降低了電解液的黏度和成本有望實現商業化。該類電解液體系舍棄了傳統穩定性較低的酯類溶劑,可以設計探索更多的電解液-鋰鹽體系,并匹配新型電極材料,有望開發出更加穩定的電池體系降低產氣。
