正交實驗優化甘草多糖膠囊中總多糖含量測定
2023-10-07 09:00:46高嘉雯朱旭江姚世霞牛鈺婷
中國民族民間醫藥 2023年16期
高嘉雯 朱旭江,* 姚世霞 牛鈺婷
1.甘肅中醫藥大學藥學院,甘肅 蘭州 730000;2.甘肅省藥品檢驗研究院,甘肅 蘭州 730070
甘草多糖膠囊系經水提醇沉法提取甘草中的多糖,加輔料制成的膠囊劑,為甘肅省中醫院的院內制劑,臨床用于提高免疫力[1-2]。該制劑的質量標準中采用3,5-二硝基水楊酸比色法(DNS),單點外標法測定甘草多糖的含量,試驗時發現操作過程復雜,試劑配制耗時,且輔料存在干擾,導致結果重現性較差。常用的多糖測定方法有:苯酚-硫酸法、蒽酮-硫酸法、DNS法等[3-5]。其中,苯酚-硫酸法操作簡便,測定快速,較為常用[6-7]。許多研究[8-10]發現,苯酚-硫酸法在測定結果的準確度和穩定性上均優于蒽酮-硫酸法。有研究[11-12]發現,在同質量濃度下,對比葡萄糖對照品溶液吸光度,苯酚-硫酸法的吸光度大于蒽酮-硫酸法和DNS法,表明苯酚硫酸法的測定結果更靈敏。因此,通過研究,建立了苯酚-硫酸比色法測定甘草多糖膠囊的含量,并通過正交實驗設計優化顯色條件,方法簡便準確,可用于該制劑的質量控制。
1 儀器與試藥
1.1 儀器 Lambda365紫外可見分光光度計(PerkinElmer公司);MS205DU十萬分之一天平(瑞士梅特勒公司);ME204電子天平(瑞士梅特勒公司);KQ-500DE數控超聲波清洗器(昆山市超聲儀器有限公司);漩渦混合器(IKA VORTEX2)。
1.2 試藥 甘草多糖膠囊(批號分別為:200402、200403、200301,甘肅省中醫院提供);D-無水葡萄糖(批號:110833-201707,含量99.9%,中國食品藥品檢定研究院)蒽酮、苯酚、3,5-二硝基水楊酸、酒石酸鉀鈉、氫氧化鈉、硫酸,均為分析純。
2 方法與結果
2.1 溶液的制備
2.1.1 對照品溶液制備 精密稱取D-無水葡萄糖對照品103.49 mg,置100 mL量瓶中,加水至刻度,搖勻,作為對照品儲備液。精密量取儲備液5 mL,置25 mL量瓶中,加水稀釋至刻度,搖勻,即得對照品溶液。
2.1.2 供試品溶液制備 取甘草多糖膠囊內容物適量,混勻,研細,精密稱取0.1 g,置250 mL量瓶中,加水200 mL,旋渦振蕩器混合5 min,超聲(功率500 W,頻率40 kHz)處理30 min,放冷至室溫,加水至刻度,搖勻,取適量,過濾,精密量取濾液5 mL,置10 mL量瓶中,用水稀釋至刻度,搖勻,即得。
2.2 最大吸收波長的測定 精密吸取0.4 mL“2.1.1”項下對照品溶液,置刻度試管中,加入5%的苯酚溶液1.0 mL,振蕩搖勻,迅速加入5 mL濃硫酸,立即搖勻,冷卻至室溫。以相應試劑為空白,在400~700 nm掃描光譜。由圖1結果可知對照品溶液在490 nm處有最大吸收,故選擇490 nm為測定波長。
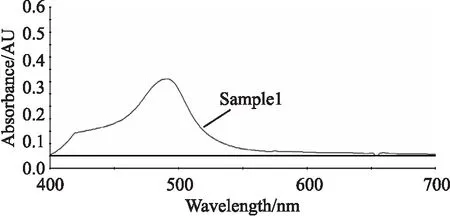
圖1 對照品溶液的可見光譜圖
2.3 正交實驗設計優化顯色條件 通過前期實驗發現,苯酚和硫酸的加入量、反應時間均對結果有較大影響,因此,采用L9(34)正交試驗設計,以吸收度為考察指標優選最佳顯色條件,并運用SPSS 27.0統計軟件對試驗結果進行方差分析。正交試驗因素與水平見表1,正交試驗設計與結果見表2,方差分析結果見表3。

表1 正交試驗因素水平表

表2 正交試驗設計與結果表
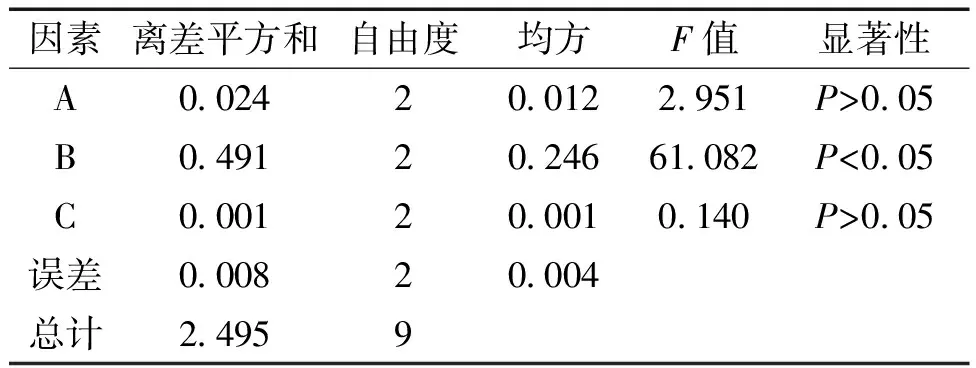
表3 方差分析結果表
由表2直觀分析可知,各因素對綜合評分影響的順序為B>A>C;由表3的方差分析可知,濃硫酸用量對顯色具有顯著意義。綜合直觀分析和方差分析結果,確定總多糖含量測定條件的最佳工藝條件為A2B1C3,即苯酚用量為1 mL,硫酸用量為5 mL,放置時間為30 min。經對同一批樣品制備3批供試品溶液進行驗證,結果RSD為3.0%(n=3),顯色條件方法可行。
2.4 標準曲線的繪制 精密吸取葡萄糖對照品溶液0.3 mL、0.4 mL、0.5 mL、0.6 mL、0.7 mL、0.8 mL,分別置具塞刻度試管中,加水至1 mL,搖勻,加入5 %苯酚溶液1 mL,搖勻,迅速加入硫酸5 mL,搖勻,放置30 min,加水至10 mL,冰浴冷卻,以相應的試劑為空白,照紫外-可見分光光度法(2020版中國藥典通則0101),在490 nm處測定吸光度。以吸光度為縱坐標、濃度為橫坐標,繪制標準曲線。回歸方程為方程為:y=0.0048x+0.0252,r=0.9995,表明葡萄糖在0.0621~0.1656 mg·mL-1內線性關系良好。
2.5 專屬性考察 按處方比例及制備工藝,制成不含甘草多糖的陰性樣品,按“2.1.2”制成供試品溶液,測定吸收度。結果吸收度小于0.001,表明輔料不干擾實驗,有較好的專屬性。
2.6 精密度試驗 精密吸取“2.1.1”項下葡萄糖對照品溶液0.4 mL,按“2.4”項下方法,重復測定吸光度6次,結果6次吸光度的RSD=0.06%(n=6),表明精密度較好。
2.7 重復性試驗 取甘草多糖膠囊(批號:200402)粉末約0.1 g,精密稱定,平行6份,按“2.1.2”制成供試品溶液,按照“2.4”項下方法測定其吸光度A,用標準曲線計算供試品溶液中葡萄糖的含量(mg/g)。結果平均含量為345.98 mg/g,RSD=1.88 %(n=6)。
2.8 穩定性試驗 取同一批號樣品(批號:200402),精密稱取0.1 g,按“2.1.2”項下方法制成供試品溶液,按照“2.4”項下方法測定其吸光度,每間隔 5 min測定1次,共測定6次。結果平均吸光光度為0.3864,RSD值為1.93 %(n=6)。表明樣品甘草多糖測定在30 min內基本穩定。
2.9 加樣回收試驗 取已知含量的甘草多糖膠囊(批號:200402)9份,每份0.05 g,精密稱定,分別精密加入其樣品多糖含量的50 %、100 %、150 %的對照品溶液,每個比例加3份,按“2.1.2”制成供試品溶液。分別精密移取 1 mL,按照測定標準曲線的方法測定其吸光度,測定平均加樣回收率為99.5%,RSD = 3.0%(n=9),表明回收率良好。結果見表4。

表4 加樣回收試驗結果表
2.10 樣品的含量 測定取3批甘草多糖膠囊各0.1 g,精密稱定,按“2.1.2”項下方法制備供試品溶液,按“2.4”項下方法測定吸光度,計算其中多糖含量。結果見表5。
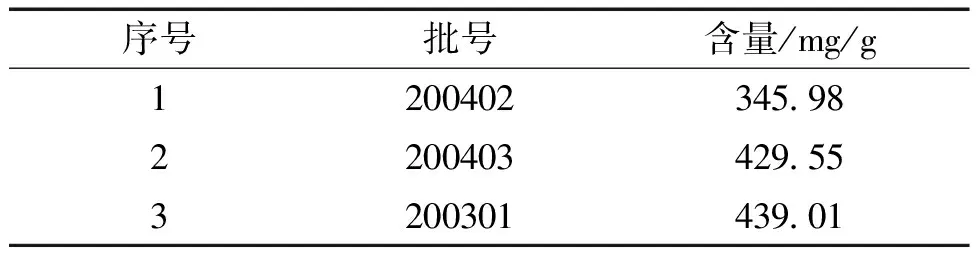
表5 樣品的含量測定結果表
3 討論
苯酚-硫酸法是利用多糖在濃硫酸作用脫水生成糠醛衍生物,糠醛與苯酚生成橙黃色化合物,再以比色法測定[13]。本試驗在單因素試驗基礎上,建立正交試驗設計,考察了苯酚、濃硫酸用量和反應時間3個因素對甘草多糖含量測定的影響。最終確顯色條件為即加入苯酚1 mL,硫酸5 mL,放置時間為30 min。該方法操作簡便,試劑易得,能快速準確測定甘草多糖的含量。
此外,分別選用超聲法、回流法、水浴保溫法以及直接溶解法等4種方式作為樣品中多糖的提取方法制備供試品溶液,在490 nm處測定吸光度值,結果顯示,超聲法測定結果較高,且操作簡便,故選用其作為樣品的制備方法。
通過方法學考察,驗證了本方法精密度高,重復性好,穩定性和準確度均較好。采用苯酚-硫酸法測定甘草多糖膠囊中多糖的含量,方法可行,操作簡單,可用于甘草多糖膠囊的質量控制,對指導臨床用藥及制訂制劑質量標準有一定參考價值。
