瑞氏紅魴(Satyrichthys rieffeli)基因組survey分析及線粒體基因組注釋*
2023-10-17 07:12:42廖賢暉王乙婷瞿印權高天翔
海洋與湖沼 2023年5期
廖賢暉 王乙婷 瞿印權 劉 啟 高天翔①
(1.浙江海洋大學水產學院 浙江舟山 316022; 2.武漢萬摩科技有限公司 湖北武漢 430076)
高通量測序已成為一種廣泛用于動植物基因組測序技術(孟剛等, 2021; 王子寅等, 2023)。通過高通量測序技術可以進行基因組survey 研究, 包括基因組大小估計、重復序列比例和GC 含量計算, 以及對遺傳多樣性、基因組結構和遺傳改良的了解(Barchiet al, 2011; Roweet al, 2011; Xuet al, 2014; Shiet al,2018), 其結果可用于調查基因組圖譜和提取線粒體基因組。對于缺乏基因組數據的非模式物種來說, 針對基因組的survey 分析是分子機理研究和基因資源開發的前提(Kimet al, 2011)。
線粒體是半自主細胞性細胞器, 普遍存在于真核細胞內(Boore, 1999)。脊椎動物的線粒體 DNA(Mitochondrial DNA, mtDNA)基因結構緊湊, 長度一般在15~20 kb 之間, 并且編碼的基因庫非常保守(張方等, 1998)。脊椎動物的線粒體基因組中一般包括了22 個tRNA 基因、2 個rRNA 基因、13 個蛋白質編碼區(PCGs)、1 個非編碼區包括D-loop 區和1 個輕鏈復制起始區(OL) (Satohet al, 2016)。線粒體基因組具有結構簡單、編碼區域高度保守、母系遺傳、進化速率快、拷貝數高等鮮明特征(Ruanet al, 2020), 被廣泛應用于群體遺傳學、進化生物學和譜系遺傳學等方面(Koet al, 2018; Zenget al, 2018; 匡衛民等, 2019;Chanet al, 2019)。線粒體物種特異性DNA 片段, 如核糖體RNA (12S rRNA 和16S rRNA)、細胞色素b(Cytb)和細胞色素c氧化酶I (COI)通常用于魚類物種鑒定(Cutarelliet al, 2018)。目前有較多的學者用線粒體基因組全序列構建系統發育樹來比較進化關系,相比于其他的COI、12S rRNA 和16S rRNA 等單一基因序列構建的系統發育樹可以減少因信息分布的不均一性和序列長度太短而造成的進化樹置信度降低帶來的影響, 使得研究結果更精確、可靠(肖家光, 2015)。
1 材料與方法
1.1 樣品采集與保存
1.2 實驗方法
1.2.1 基因組survey 測序 用標準的苯酚-氯仿法提取肌肉組織的總基因組DNA (Maniatiset al, 1982)。檢測合格的DNA 樣本通過超聲波破碎儀隨機打斷成長度為300~350 bp 的片段, 經末端修復、加A 尾、加測序接頭、純化、PCR 擴增等步驟完成整個文庫制備。構建好的文庫通過Illumina nova 進行PE150 測序。使用fastp 軟件對測序數據進行數據過濾與去冗余操作(Kajitaniet al, 2014), 過濾后得到的高質量數據將用于后續的雜合度、GC 含量以及基因組大小等信息分析。
1.2.2 基因組大小預估和K-mer 分析 利用過濾后的數據, 取K=17, 參照Liu 等(2013)的K-mer 分析方法對瑞氏紅魴基因組大小、雜合率和重復率等基因特征進行進一步分析。基于K-mer 分析的結果, 獲得了關于峰的深度和預測的最佳K-mer 數量的信息,并用于估計基因組的大小。
1.2.3 基因組組裝 使用SOAPdenovo 軟件(Luoet al, 2012)對過濾去除低質量數據之后的高質量測序數據進行基因組預組裝分析。采用K=48 構建Contig和Scaffold, 利用高質量數據進行全基因組裝, 使用SOAPdenovo 軟件將過濾后的reads 比對拼接到該組裝序列上, 獲得原始基因組序列及堿基深度。
1.2.4 線粒體基因組組裝和注釋 借助NOVOPlasty 2.6.3 軟件從原始的基因組測序數據中提取線粒體基因組序列(Dierckxsenset al, 2017), 參數采用默認設置, 參考序列為GenBank 瑞氏紅魴COI 序列(GenBank No.MK777566.1)。通過MitoFish將得到的數據進行注釋和可視化(Iwasakiet al, 2013)。利用MEGA 11 計算基因組堿基組成? 氨基酸使用頻率及同義密碼子相對使用頻率(匡衛民等, 2019)。
1.2.5 系統進化分析 GenBank 中下載18 種 鲉形目魚類的線粒體基因組, 以豹魴(Dactylopterus volitans)為外群, 基于12 種蛋白質編碼基因(除ND6)進行系統發育分析, 確定瑞氏紅魴的分類地位。利用MEGA X 和貝葉斯法(Bayesian)構建最大似然樹(ML)和貝葉斯樹(BI) (Ronquistet al, 2003), 采用MEGA X 方法估計了最優核苷酸替換模型(Kumaret al, 2018), 基于BIC 標準得到最優GTR+G+I 模型, 確定構建系統發育樹的最佳模型。利用MEGA X 軟件構建了1 000 個重復的最大似然系統發育樹, 探究系統進化關系?
2 結果
2.1 基因組survey 結果
2.1.1 測序數據統計及質量評估 使用Illumina nova 測序平臺進行雙末端測序, 使用瑞氏紅魴基因組總DNA 構建300 bp 的文庫, 測序得到raw reads 57.59 Gb, 質控后得到約49.13 Gb 的clean reads。瑞氏紅魴質控后其Q20值為96.73%,Q30值為91.72%(表1), 顯示本次瑞氏紅魴的高通量測序結果有較高的準確性, 結果可用于后續的分析。
表1 瑞氏紅魴survey 測序結果Tab.1 Results of the survey sequencing of S. rieffeli
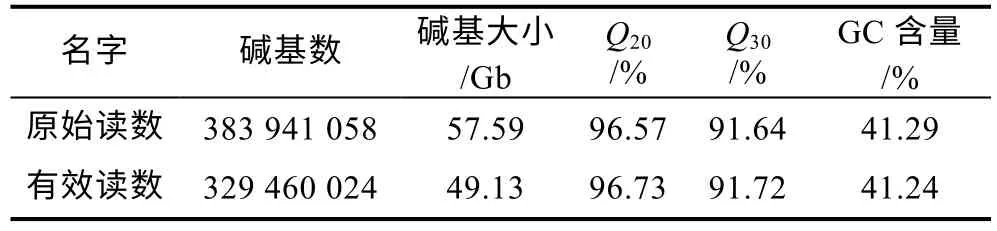
表1 瑞氏紅魴survey 測序結果Tab.1 Results of the survey sequencing of S. rieffeli
注: Q20: 質量百分比≥20; Q30: 質量百分比≥30
名字 堿基數 堿基大小/Gb Q20/%Q30/%GC 含量/%原始讀數 383 941 058 57.59 96.57 91.64 41.29有效讀數 329 460 024 49.13 96.73 91.72 41.24
表2 瑞氏紅魴高通量測序結果與NT 庫比對Tab.2 The results of S. rieffeli comparision with the NT library
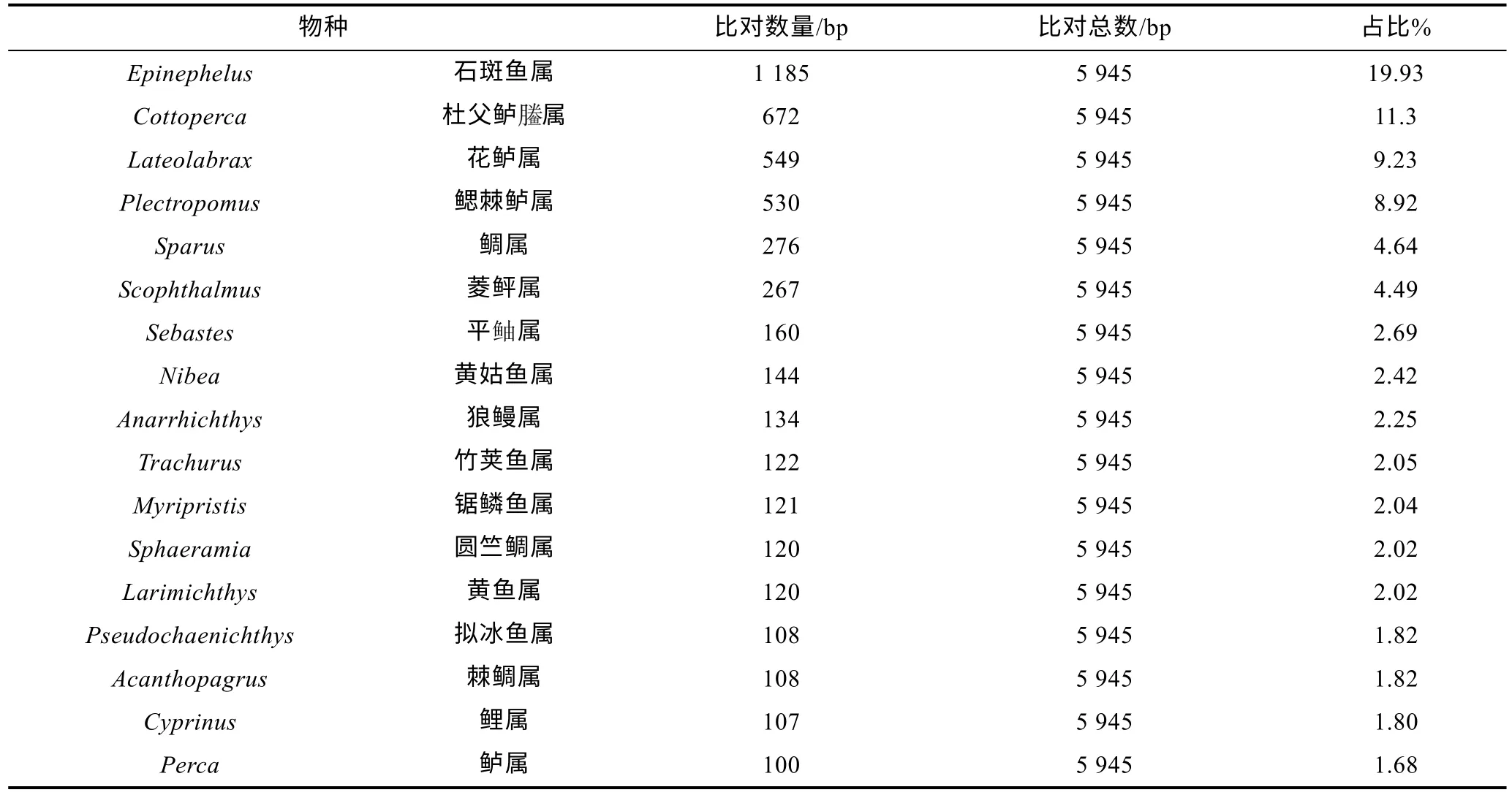
表2 瑞氏紅魴高通量測序結果與NT 庫比對Tab.2 The results of S. rieffeli comparision with the NT library
物種 比對數量/bp 比對總數/bp 占比%Epinephelus 石斑魚屬 1 185 5 945 19.93 Cottoperca 杜父鱸images/BZ_285_912_795_944_826.png屬 672 5 945 11.3 Lateolabrax 花鱸屬 549 5 945 9.23 Plectropomus 鰓棘鱸屬 530 5 945 8.92 Sparus 鯛屬 276 5 945 4.64 Scophthalmus 菱鲆屬 267 5 945 4.49 Sebastes 平鲉 屬 160 5 945 2.69 Nibea 黃姑魚屬 144 5 945 2.42 Anarrhichthys 狼鰻屬 134 5 945 2.25 Trachurus 竹莢魚屬 122 5 945 2.05 Myripristis 鋸鱗魚屬 121 5 945 2.04 Sphaeramia 圓竺鯛屬 120 5 945 2.02 Larimichthys 黃魚屬 120 5 945 2.02 Pseudochaenichthys 擬冰魚屬 108 5 945 1.82 Acanthopagrus 棘鯛屬 108 5 945 1.82 Cyprinus 鯉屬 107 5 945 1.80 Perca 鱸屬 100 5 945 1.68
2.1.3 K-mer 分析 使用K-mer 分析預測了瑞氏紅魴的基因組特征及雜合率和重復比例。選取K=17, 預測瑞氏紅魴基因組大小為827 Mb, 修正后的基因組大小為813 Mb; 基因組雜合率為0.92%,重復序列比例為45.97% (表3)。其K-mer 深度在48時出現一個期望深度, 且沒有出現其他峰值(圖1),說明瑞氏紅魴基因組雜合度較低。
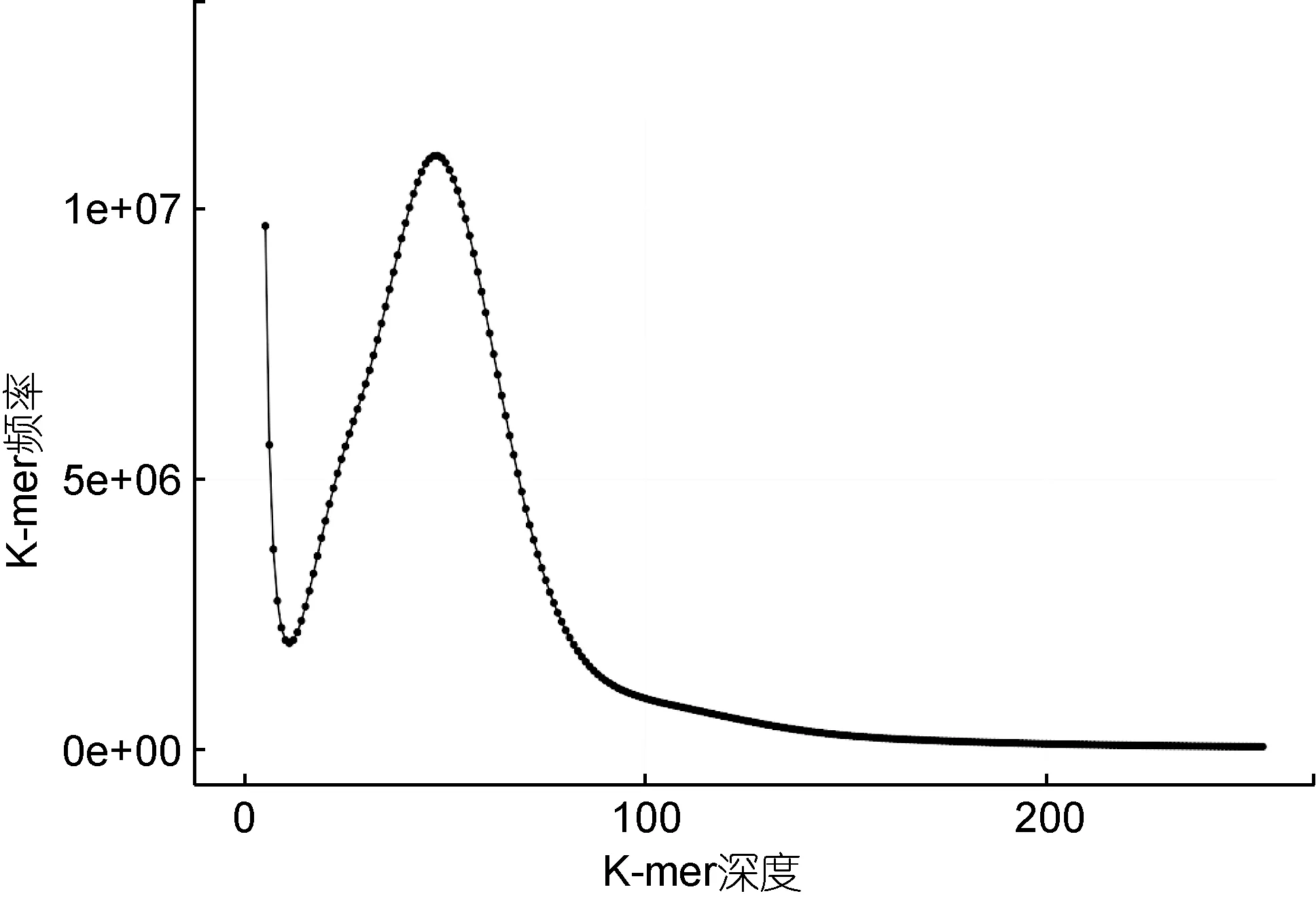
圖1 瑞氏紅魴K-mer 深度分布圖Fig.1 Distribution of the K-mer depth of S. rieffeli
表3 瑞氏紅魴基因組特征統計Tab.3 Statistics of S. rieffeli genome characteristics

表3 瑞氏紅魴基因組特征統計Tab.3 Statistics of S. rieffeli genome characteristics
樣品 K-mer 數 K-mer 深度 基因組大小/Mb 修正后基因組大小/Mb 雜合率/% 重復率/%S. rieffeli 43 850 343 050 48 827 813 0.92 45.97

表4 Contigs 組裝結果統計Tab.4 Statistics of the contigs assembly
2.2 線粒體基因組注釋
2.2.1 線粒體基因組組成及其特征 從survey 結果里面提取并組裝線粒體基因組, 結果顯示瑞氏紅魴的線粒體全序列長16 527 bp, 其中有38 個基因獲得注釋, 分別由22 個tRNA、13 個蛋白質編碼基因(PCGs:APT6、ATP8、Cyt-b、COXI~COXIII、ND1~ND6和ND4L)、2 個rRNA (12S rRNA和16S rRNA)和1 個D-loop 區組成(圖2, 表5)。
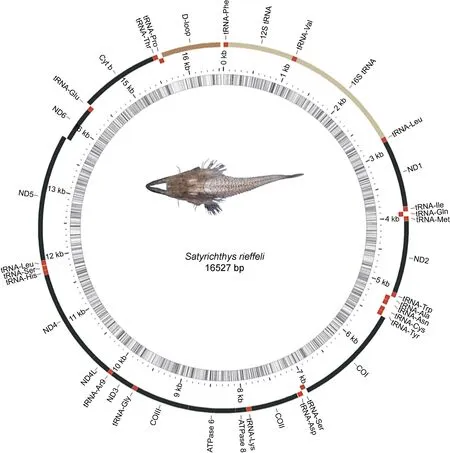
圖2 瑞氏紅魴線粒體基因組的圓形圖譜Fig.2 The circular map of the mitogenome of S. rieffeli
表5 瑞氏紅魴的線粒體基因組結構特征Tab.5 Structural characteristics of the mitochondrial genome of S. rieffeli
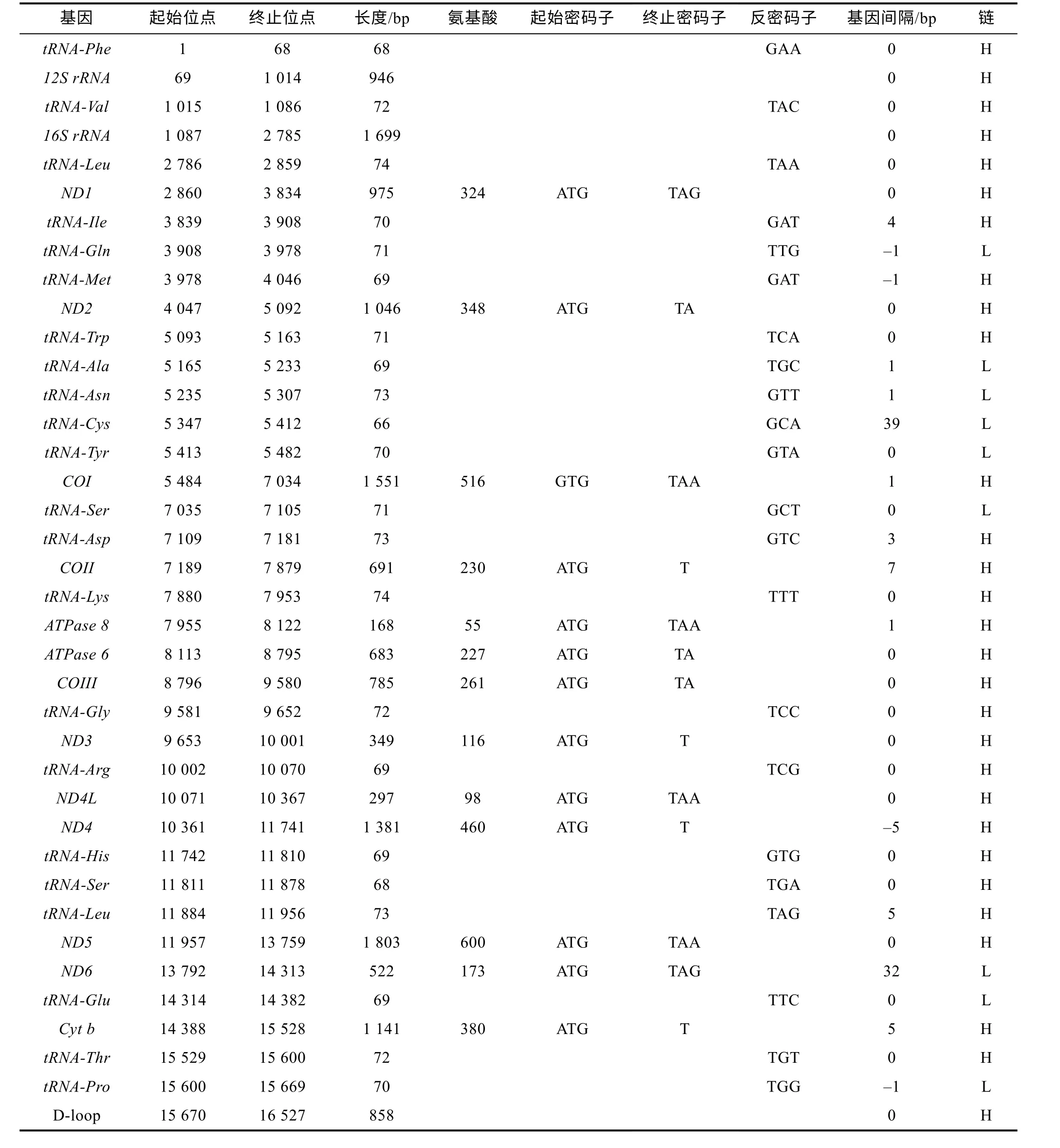
表5 瑞氏紅魴的線粒體基因組結構特征Tab.5 Structural characteristics of the mitochondrial genome of S. rieffeli
點 終止位點 長度/bp 氨基酸 起始密碼子 終止密碼子 反密碼子 基基因 起始位 因間隔/bp 鏈tRNA-Phe 1 68 68 GAA 0 H 12S rRNA 69 1 014 946 0 H tRNA-Val 1 015 1 086 72 TAC 0 H 16S rRNA 1 087 2 785 1 699 0 H tRNA-Leu 2 786 2 859 74 TAA 0 H ND1 2 860 3 834 975 324 ATG TAG 0 H tRNA-Ile 3 839 3 908 70 GAT 4 H tRNA-Gln 3 908 3 978 71 TTG –1 L tRNA-Met 3 978 4 046 69 GAT –1 H ND2 4 047 5 092 1 046 348 ATG TA 0 H tRNA-Trp 5 093 5 163 71 TCA 0 H tRNA-Ala 5 165 5 233 69 TGC 1 L tRNA-Asn 5 235 5 307 73 GTT 1 L tRNA-Cys 5 347 5 412 66 GCA 39 L tRNA-Tyr 5 413 5 482 70 GTA 0 L COI 5 484 7 034 1 551 516 GTG TAA 1 H tRNA-Ser 7 035 7 105 71 GCT 0 L tRNA-Asp 7 109 7 181 73 GTC 3 H COII 7 189 7 879 691 230 ATG T 7 H tRNA-Lys 7 880 7 953 74 TTT 0 H ATPase 8 7 955 8 122 168 55 ATG TAA 1 H ATPase 6 8 113 8 795 683 227 ATG TA 0 H COIII 8 796 9 580 785 261 ATG TA 0 H tRNA-Gly 9 581 9 652 72 TCC 0 H ND3 9 653 10 001 349 116 ATG T 0 H tRNA-Arg 10 002 10 070 69 TCG 0 H ND4L 10 071 10 367 297 98 ATG TAA 0 H ND4 10 361 11 741 1 381 460 ATG T –5 H tRNA-His 11 742 11 810 69 GTG 0 H tRNA-Ser 11 811 11 878 68 TGA 0 H tRNA-Leu 11 884 11 956 73 TAG 5 H ND5 11 957 13 759 1 803 600 ATG TAA 0 H ND6 13 792 14 313 522 173 ATG TAG 32 L tRNA-Glu 14 314 14 382 69 TTC 0 L Cyt b 14 388 15 528 1 141 380 ATG T 5 H tRNA-Thr 15 529 15 600 72 TGT 0 H tRNA-Pro 15 600 15 669 70 TGG –1 L D-loop 15 670 16 527 858 0 H
除8 個tRNA 基因(tRNA-Gln、Ala、Asn、Cys、Tyr、Ser、Glu、Pro)和1 個PCGs 基因(ND6)在線粒體基因組的L 鏈上, 其他29 個基因和D-loop 區均在H 鏈上, 基因間都未發生重排。其中11 個基因發生了間隔, 共99 bp,tRNA-Cys基因間隔最長, 共39 bp,其次是基因ND6, 共32 bp; 有4 個基因發生了重疊,共8 bp, 重疊的基因是tRNA-Gln、tRNA-Met、ND4和tRNA-Pro, 其中基因ND4 重疊最多, 有5 bp。瑞氏紅魴線粒體全基因堿基組成: A (27.4%)、T(26.9%)、G (16.8%)、C (28.9%) (表6), 各基因的GC含量在44.4%~51.4%之間, 其中A+T 的含量(54.3%)大于G+C 的含量(45.7%)。
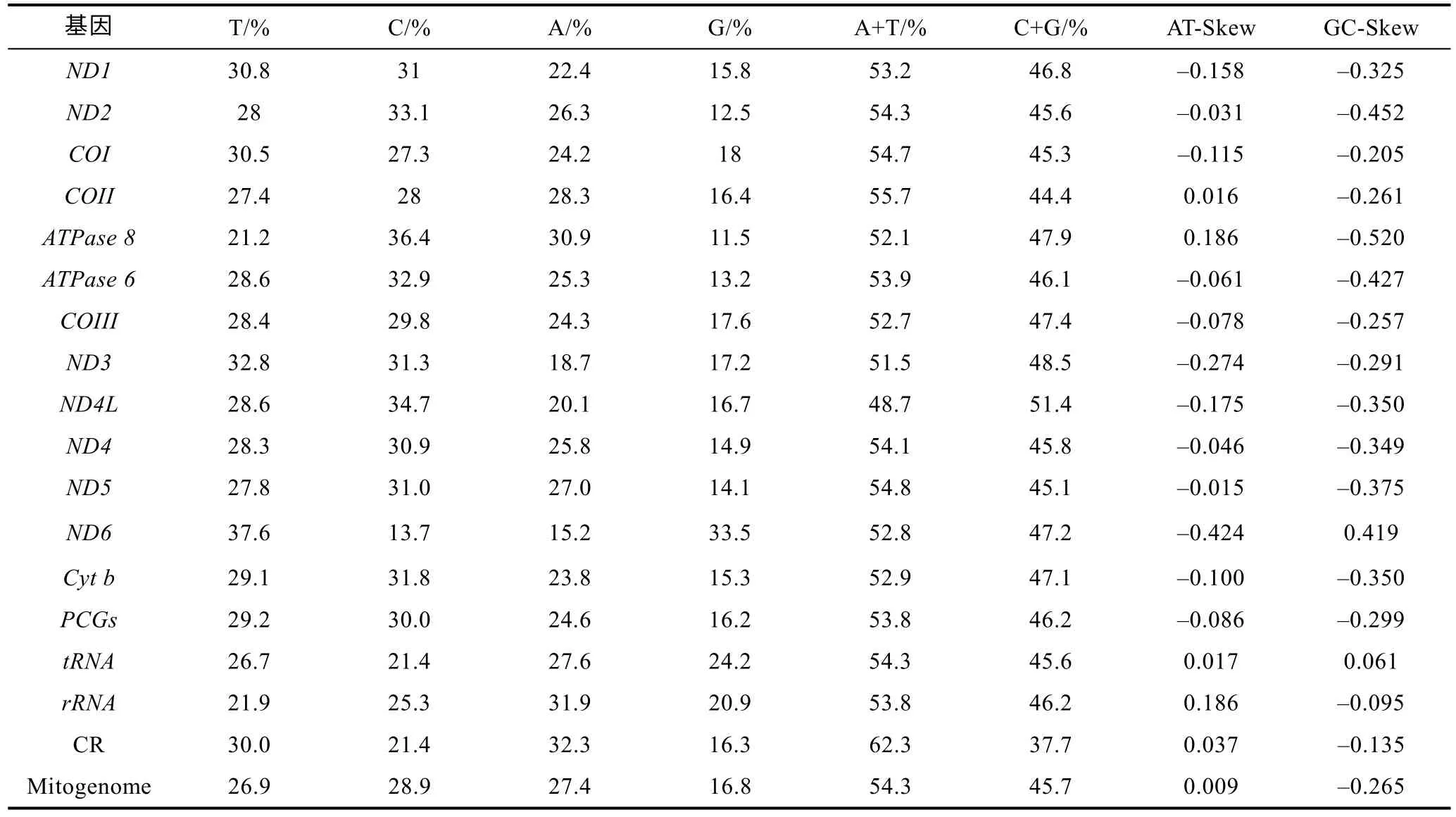
表6 線粒體基因組核苷酸組成比例Tab.6 The composition ratio of mitochondrial genome nucleotide
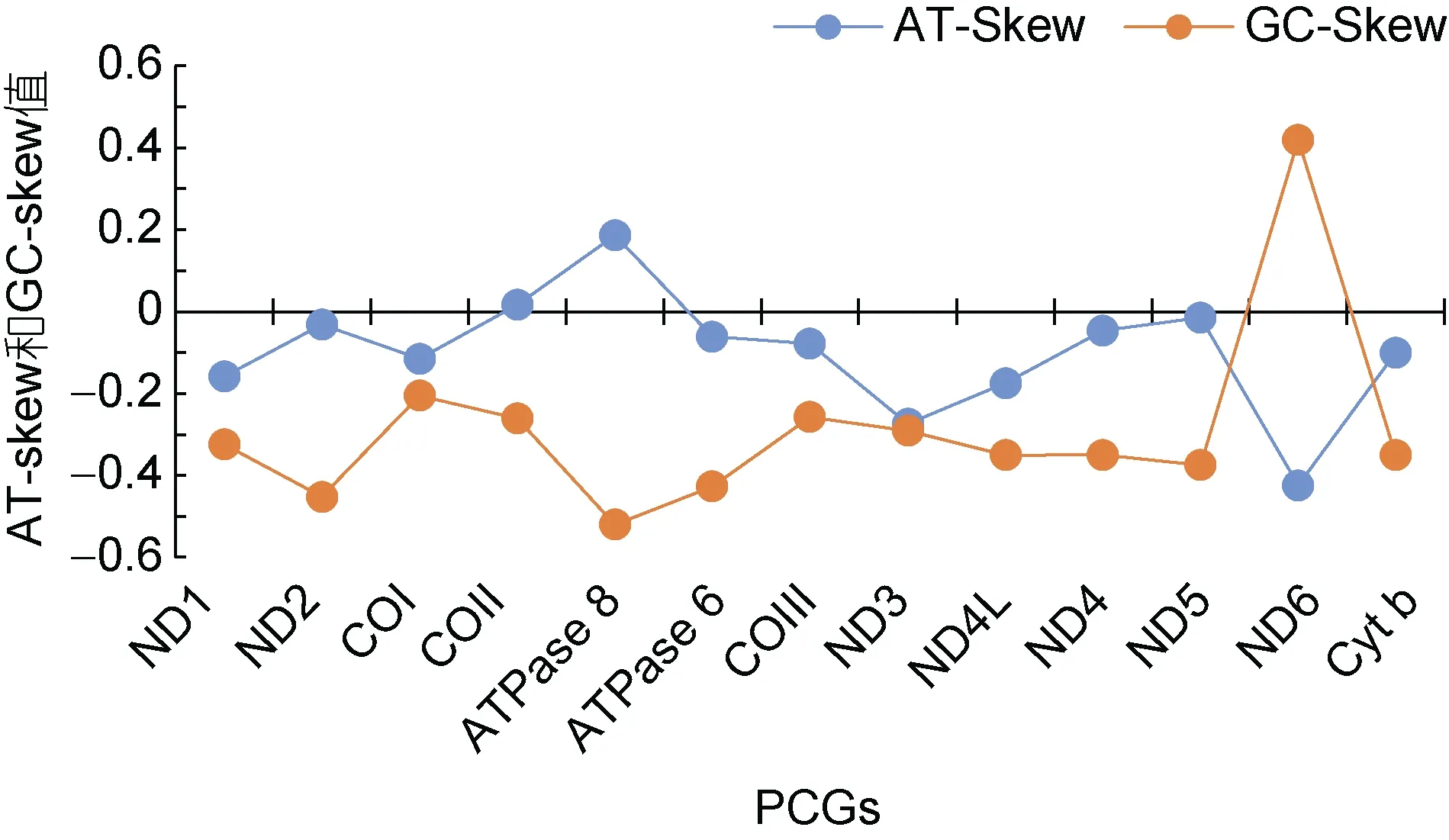
圖3 瑞氏紅魴13 個蛋白質編碼基因的AT-skew 和GC-skew 值Fig.3 The AT-skew and GC-skew values of 13 protein-coding genes in S. rieffeli
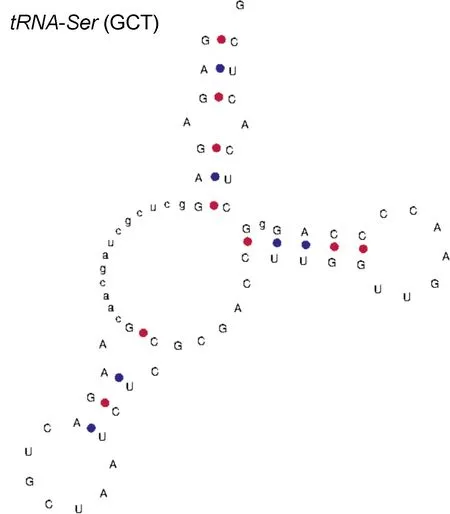
圖4 瑞氏紅魴tRNA-Ser (GCT)二級結構預測圖Fig.4 Prediction of secondary structure of tRNA-Ser (GCT) in S.rieffeli

圖5 瑞氏紅魴線粒體基因密碼子使用頻率Fig.5 Frequency of usage of mitochondrial gene codon of S. rieffeli
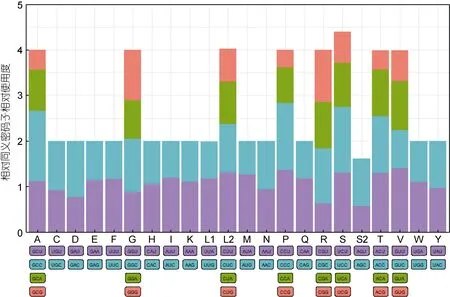
圖6 瑞氏紅魴線粒體基因相對同義密碼子使用頻率Fig.6 Frequency of relative synonymous codon usage (RSCU) in S. rieffeli
2.2.5 非編碼區 非編碼區又稱之為 D-環區(displacement-loop region), 位 于tRNA-Pro和tRNA-Phe基因之間, 是整個線粒體基因組序列和長度變異最大的區域, 但其中也包含保守片段。瑞氏紅魴的非編碼區各堿基含量分別為T (30.0%)、C (21.4%)、A (32.3%)、G (16.3%), 表現出明顯的AT 偏好性, A+T的含量達到了62.3%, G+C 的含量僅為37.7%。
2.3 系統發育關系
3 討論
近年來, 高通量測序技術的迅速發展為解決非模式海洋魚類的基因組問題提供了有效的技術手段。利用K-mer 方法可以估計非模型物種的基因組大小,無須任何先驗知識(Liet al, 2019b), 其方法已被應用于絨杜父魚(Hemitripterus villosus) (趙蕊蕊等, 2022)、褐菖 鲉 (Sebastiscus marmoratus) (Xuet al, 2020)、菖鲉屬(Sebastiscus) (Jiaet al, 2021) 等 鲉形目魚類的研究。目前瑞氏紅魴的分子信息還不完善, 本研究survey 結果顯示GC 含量在41%左右出現峰值(雙端測序結果都出現在41%左右), 且沒有出現明顯的偏差, 表明測序結果沒有偏向性; 并獲得49.13 Gb 的clean reads,Q20大于90%, 基因組大小為827 Mb, 修正后的基因組大小為813 Mb, 其雜合率為0.92%, 重復率為45.97%。瑞氏紅魴與大多數的海洋魚類相似, 其基因組大小稍大于絨杜父魚713.18 Mb (趙蕊蕊等, 2022) 、褐菖 鲉812.86 Mb (Xuet al, 2020)、艾氏蛇鰻(Ophichthus lithinus) 755.24 Mb (寧子君等,2022), 稍小于許氏平鲉 (Sebastes schlegelii) 846.36 Mb、朝鮮平 鲉(Sebastes koreanus) 832.53 Mb 、金斑平鲉(Sebastes nudus) 813.12 Mb (Xuet al, 2019)、雙帶縞虎(Tridentiger bifasciatus) 887.60 Mb (Zhaoet al,2022), 導致物種間基因組大小的差異可能是由于重復序列含量差異所導致。瑞氏紅魴的重復率為45.97%, 較絨杜父魚38.61%、 褐菖 鲉39.65%、艾氏蛇鰻43.30%、 許氏平 鲉44.10%、 朝鮮平 鲉43.65%、金斑平 鲉41.40%、雙帶縞虎32.60%均較大。此外,瑞氏紅魴的雜合率為0.92%, 較絨杜父魚0.26%、褐菖 鲉0.17%、艾氏蛇鰻0.70%、 許氏平 鲉0.22%、朝鮮平 鲉0.20%、 金斑平 鲉0.31%、雙帶縞虎0.47%均較大, 一般來說, 雜合率大于0.8%, 重復率大于60%就稱為復雜基因組(高勝寒等, 2018), 由于瑞氏紅魴的雜合率高于0.8%, 重復率低于60%, 其基因組不屬于復雜基因組類型。在本研究中, 組裝的初步結果顯示Contigs 的N50 為712 bp, N50 數量為254 317, 其結果滿足基因組組裝要求。通過過濾后的clean reads 組裝瑞氏紅魴的基因組, 這是目前第一個基于二代測序技術組裝瑞氏紅魴的基因組, 該基因組為瑞氏紅魴的進化生物學研究提供了基礎數據, 也為進一步探索該物種的基因組特征提供了參考。由于二代測序技術存在讀長短、組裝結果的連續性無法保證以及由于基因組的雜合導致過度組裝或組裝不徹底等問題, 因此本研究缺少對GC-depth和含量的相關分析, 后續開展三代測序以獲得瑞氏黃魴高質量的基因組。
本研究組裝出來的線粒體基因組大小為16 527 bp,線粒體結構未出現基因重排現象, GC 含量為45.7%,出現明顯的AT 偏倚, 其結果符合硬骨魚類線粒體堿基組成偏好于堿基A 和T 的特點(Broughtonet al,2001; 蒙子寧等, 2004; Consuegraet al, 2015), 堿基G的含量為16.8%, 表現出明顯的反G 偏倚(孟剛等,2021), 與大多數魚類研究結果一致(丁少雄等,2006)。13 個PCGs 中,COI是以GTG 作為起始密碼子, 其余均為ATG 起始密碼子, 其中密碼子ATG 是翻譯效率最高的一個(Consuegraet al, 2015)。PCGs終止密碼子中出現T--和TA-這類不完整的終止密碼子, 是因為這些基因之后是一個編碼在同一條鏈上的基因, 允許轉錄在沒有完整密碼子的情況下終止(Hechtet al, 2017)。不完全終止密碼子的存在在魚類的線粒體基因組中很常見, 可以通過在mRNA 加工過程中添加一個poly A 尾巴來完成(Ojalaet al, 1981;Liuet al, 2009)。PCGs 的AT-skew 和GC-skew 為負值(表6), 一般來說, AT 傾斜的幅度小于GC 傾斜, 而且在許多情況下, 沒有統計學意義, 在這里, AT-skew 低于GC-skews (絕對值), 這符合傳統的偏好(Yuet al,2019)。AT-skew 的最大值和GC-skew 的最小值均出現在ND6 中, 且該基因的AT/GC-skew 值波動較大,核苷酸偏斜可能是由于在復制和轉錄過程中突變壓力和選擇壓力之間的平衡, 為基因復制提供了一個潛在的方向(McLeanet al, 1998; Touchonet al, 2008)。tRNA 基因結構中僅tRNA-Ser(TCG)基因缺失DHU臂的結構, 這是魚類線粒體基因組的共同特征, DHU臂的缺失改變了tRNA-Ser(TCG)在線粒體基因中的識別潛力, 使其更容易被識別(Hardtet al, 1993)。瑞氏紅魴線粒體基因組的12S rRNA和16S rRNA基因定位于H 鏈tRNA-Phe和tRNA-Leu(UUR)基因之間,中間以tRNA-Val基因為間隔,12S rRNA基因比16S rRNA基因更保守(Satohet al, 2016)。當RSCU 值=1時, 表示密碼子的使用頻率與其他簡并密碼子沒有差異; 當氨基酸的RSCU 值均不等于1, 說明每個氨基酸的使用都有不同程度的偏倚(周丹等, 2013; 孟乾等, 2020)。D-loop 區在tRNA-Pro和tRNA-Phe之間, 與其他大多數脊椎動物的排列一樣(Weiet al, 2010; Liet al, 2019a)。
目前通過線粒體基因組構建系統發育樹來確定物種的進化關系已經成為系統發育分析的常用方法,比如周志雄等(2020)通過線粒體基因組構建系統發育樹得出東方 鲀屬內系統發育關系較為復雜, 且存在野生環境下種間自然雜交現象; Miya 等(2015)通過過去15 年線粒體基因組發展應用做出的總結, 認為線粒體基因組分析和高通量測序技術會成為物種分類重要方法。本研究基于12 個PCGs 構建系統發育樹,以豹魴作為外群, 分析瑞氏紅魴的系統發育關系。ML 樹和BI 樹結果表明鲉科在ML 樹中與外群遺傳距離較近, 聚在一起, 與在BI 樹中結果不一致,但是兩個樹結果都顯示 鲉科與其他科遺傳距離都較遠。瑞氏紅魴與須叉吻魴聚為一支, 作為黃魴科魚類, 與形態學研究結果一致(Kawai, 2008)。
4 結論
致謝 感謝趙宸楓在采集樣品和鑒定方面的幫助及楊天燕老師的修改意見, 謹致謝忱。
