杏仁核N-甲基-D-天冬氨酸受體在芬太尼誘發痛覺過敏大鼠中的作用
2023-11-07 01:34:52尹平平許睿周張胡曉榆于明良
中國老年學雜志 2023年21期
關鍵詞:研究
尹平平 許睿 周張 胡曉榆 于明良
(武漢市第四醫院麻醉科,湖北 武漢 430034)
芬太尼是人工合成的阿片受體激動劑,與其他阿片類藥物一樣,具有強效的鎮痛作用,但重復使用會引起機體對疼痛的敏感性增強,即阿片誘發的痛覺過敏(OIH)〔1〕,這種副作用嚴重干擾了臨床醫生對疼痛的管理和治療〔2〕,亟待探明其發生機制。N-甲基-D-天冬氨酸受體(NMDAR)廣泛存在于中樞神經系統,包括杏仁核(CeA),且NMDAR與疼痛的發生密切相關〔3〕。研究表明,脊髓NMDAR可被細胞外調節蛋白激酶(ERK)激活,使NMDAR磷酸化增加,導致其功能增強,最終參與炎性OIH的產生〔4〕,而下調脊髓NMDAR的表達可以減弱瑞芬太尼誘導的OIH〔5〕。研究證明,CeA內ERK與OIH的產生有關〔6,7〕,但CeA內NMDAR在OIH中的作用尚待進一步研究。因此,本研究擬探討CeA內NMDAR在芬太尼誘發的OIH大鼠中的作用,以便為臨床預防和治療OIH提供思路。
1 材料與方法
1.1動物選擇 健康清潔級(SPF級)雄性SD大鼠36只,體質量70~110 g,由華中科技大學同濟醫學院附屬同濟醫院動物實驗中心提供,室溫飼養,自由攝食和飲水,白天與黑夜時間12 h∶12 h。本實驗所用的大鼠均嚴格遵循中國衛生機構制定的實驗室動物應用指南。
1.2實驗分組 實驗一:采用隨機數字表法,將24只SD大鼠隨機分為3組(n=8):芬太尼+氯胺酮組(K1組)皮下注射芬太尼制備OIH模型,1 d后腹腔注射NMDAR拮抗劑氯胺酮(15 mg/kg);芬太尼誘發OIH組(H1組)皮下注射芬太尼制備OIH模型,1 d后腹腔注射等容量生理鹽水;對照組(C1組)皮下注射等容量生理鹽水,1 d后腹腔注射等容量生理鹽水。分別于皮下注射芬太尼或生理鹽水前(T0)、注射后1 d(T1)和腹腔注射氯胺酮或生理鹽水后30 min(T2)時檢測大鼠機械痛閾和熱痛閾。實驗二:將實驗一最后一次測痛結束的各組大鼠處死,取右側CeA區組織,Western印跡檢測NMDAR亞基NR2B磷酸化(P-NR2B)的表達。實驗三:另取12只雄性SD大鼠,隨機分為3組(n=4):對照組(C2組)、芬太尼誘發OIH組(H2組)和芬太尼+氯胺酮組(K2組)按實驗一處理后,制備含有中央CeA的腦片,采用CeA區神經元電生理記錄各組右側CeA區神經元NMDAR微小興奮性突觸后電流(mEPSCs)的幅值和頻率。
1.3芬太尼誘發OIH模型的制備 枸櫞酸芬太尼(湖北宜昌人福藥業有限公司,批號1130411)使用前用生理鹽水配制。采用文獻〔8〕的方法制備芬太尼誘導的OIH模型。具體操作是:大鼠頸背部皮下注射芬太尼,每次60 μg/kg,共4次,給藥間隔15 min,累積藥量240 μg/kg。給藥期間及給藥后2 h內注意給大鼠保暖及吸氧。
1.4疼痛行為測定 分別于T0、T1和T2時測定大鼠的機械痛閾和熱痛閾。測定機械痛閾時,具體方法是將大鼠置于金屬籠內,靜置30 min后,用不同壓力的von Frey絲(North Coast公司,美國)垂直刺激大鼠左足掌面,力度以von Frey絲輕微彎曲為準,持續10 s或直至出現縮足反應。當大鼠在規定時間內出現縮足反應,則記為陽性。采用up-and-down方法計算50%縮足反應閾即為機械痛閾值。測定熱痛閾時,采用的熱痛刺激儀(BME-410C)購于中國醫學科學生物醫學工程研究所。具體方法是將大鼠置于底部為玻璃板的透明小室中,安靜放置30 min后,使用輻射燈對準大鼠左足掌正中照射,自動記錄大鼠的縮足潛伏期作為熱痛閾,停止時間為15 s,以免造成大鼠左足掌組織損傷。
1.5Western印跡檢測CeA組織P-NR2B的表達 測痛完畢后處死大鼠,并取出右側CeA區組織凍存于-80 ℃冰箱備用。取適量組織稱重后,加入一定量含有蛋白酶抑制劑的RIPA裂解液(碧云天,中國)并勻漿,12 000 r/min,4 ℃離心10 min取上清,二喹啉甲酸(BCA)法測蛋白濃度。加入十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)上樣緩沖液(碧云天,中國),95 ℃煮沸8~10 min使蛋白變性,制備SDS-PAGE(10%),電泳后濕轉至聚偏氟乙烯(PVDF)膜上。5%脫脂奶粉TBST緩沖液室溫震蕩封閉1 h后洗膜,加入封閉液稀釋的兔抗P-NR2B抗體 (1∶500稀釋,Millipore公司,德國)和小鼠抗GAPDH(1∶500,博士德,中國),4 ℃孵育過夜,洗膜,加入TBST稀釋的辣根過氧化酶(HRP)-山羊抗兔和HRP-山羊抗小鼠的二抗(1∶5 000),室溫孵育2 h,洗膜后在避光條件下將電化學發光(ECL)試劑盒中的A、B兩種試劑等體積混合,振蕩混勻后均勻加到膜上,用化學發光凝膠圖像系統拍照。P-NR2B表達量以P-NR2B與GAPDH光密度的比值表示。
1.6全細胞膜片鉗實驗 全細胞膜片鉗技術記錄大鼠CeA區神經元NMDAR mEPSCs。采用LEICA VT1000 S切片機,將含有CeA區的大鼠右腦組織塊冠狀切成厚度為350 μm的腦片。切片液成分(mmol/L):213蔗糖,3 KCl,1 NaH2PO4,0.5 CaCl2,5 MgCl2,26 NaHCO3和10葡萄糖,切片液溫度為4 ℃。將切片所得的含有CeA區的腦片在25 ℃的人工腦脊液(ACSF)中孵育,時間至少1 h,人工ACSF成分(mmol/L):125 NaCl,5 KCl,1.2 NaH2PO4,2.6 CaCl2,1.3 MgCl2,26 NaHCO3和10葡萄糖。其中切片液及人工ACSF均為氧飽和溶液,且調節PH至7.2~7.4,滲透壓維持在290~310 mOsm。使用的持續灌流ACSF的溫度控制在31 ℃,灌流速度為2 ml/min。膜片鉗放大器為HEKA EPC-10 (Molecular Devices),記錄軟件為patchmaster (Molecular Devices),應用紅外線微分干涉相差顯微鏡在可視化模式下記錄CeA區神經元mEPSCs。確認封接電阻(>2 GΩ)和串聯電阻(<20 MΩ)以保證細胞封接質量。記錄電極為玻璃微電極(WPI),電極內液成份(mmol/L):100葡萄糖酸鉀、30 KCl、5 NaCl、1 MgCl2、10 HEPES、 3 EGTA、2 Na2-ATP,用1 mmol/L NaOH調節PH值為7.4。持續灌流ACSF中加入 50 μmol/L木防已苦毒素(PTX)阻斷抑制性突觸后電流,1 μmol/L河豚毒素(TTX,阻斷動作電位)和20 μmol/L CNQX(AMPA受體和海人藻受體的拮抗劑),且鉗制電壓+60 mV的條件下進行CeA區神經元細胞的高阻封接,從而獲得NMDA受體mEPSCs。
1.7統計學方法 采用SPSS21.0軟件進行方差分析、t檢驗。
2 結 果
2.1芬太尼誘發的OIH可以被NMDAR拮抗劑所逆轉 H1組T1、T2時及K1組T1時機械縮足閥值和熱縮足潛伏期均明顯低于本組T0時(均P<0.05);與C1組比較,H1組T1、T2時、K1組T1時機械縮足閾值和熱縮足潛伏期均明顯降低(P<0.05);而給予氯胺酮后可逆轉OIH,即與H1組比較,K1組T2時機械痛閾和熱縮足潛伏期均明顯升高(均P<0.05),見表1。
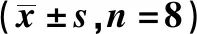
表1 各組不同時間點機械縮足閾值和熱縮足潛伏期比較
2.2芬太尼誘發的OIH與大鼠CeA中P-NR2B的過度表達有關 與C1組CeA區P-NR2B水平(0.28±0.01)相比,H1組(0.48±0.05)明顯增多(P<0.05),K1組(0.28±0.02)差異無統計學意義(P>0.05)。見圖1。
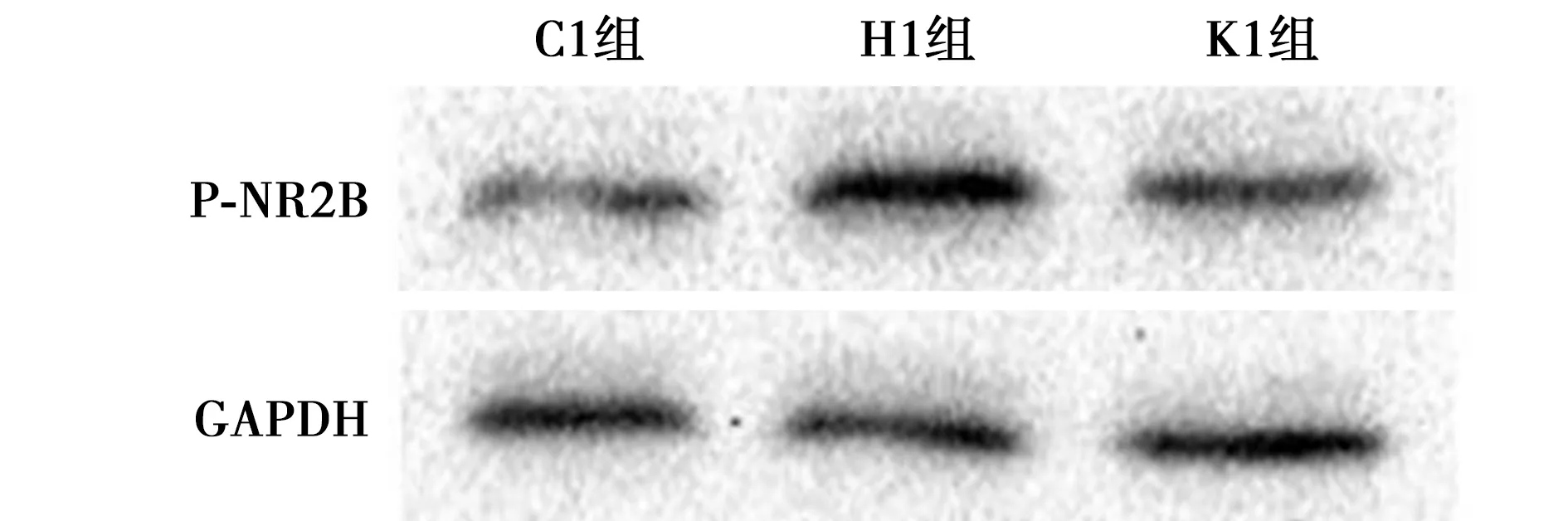
圖1 Western印跡檢測3組P-NR2B水平
2.3芬太尼誘發的OIH大鼠CeA區神經元NMDAR介導的突觸傳遞發生改變 CeA區神經元電生理記錄示,與C2組比較,H2組CeA區NMDAR mEPSCs幅值及頻率均明顯增加(均P<0.05),而K2組差異均無統計學意義(均P>0.05)。見表2、圖2。
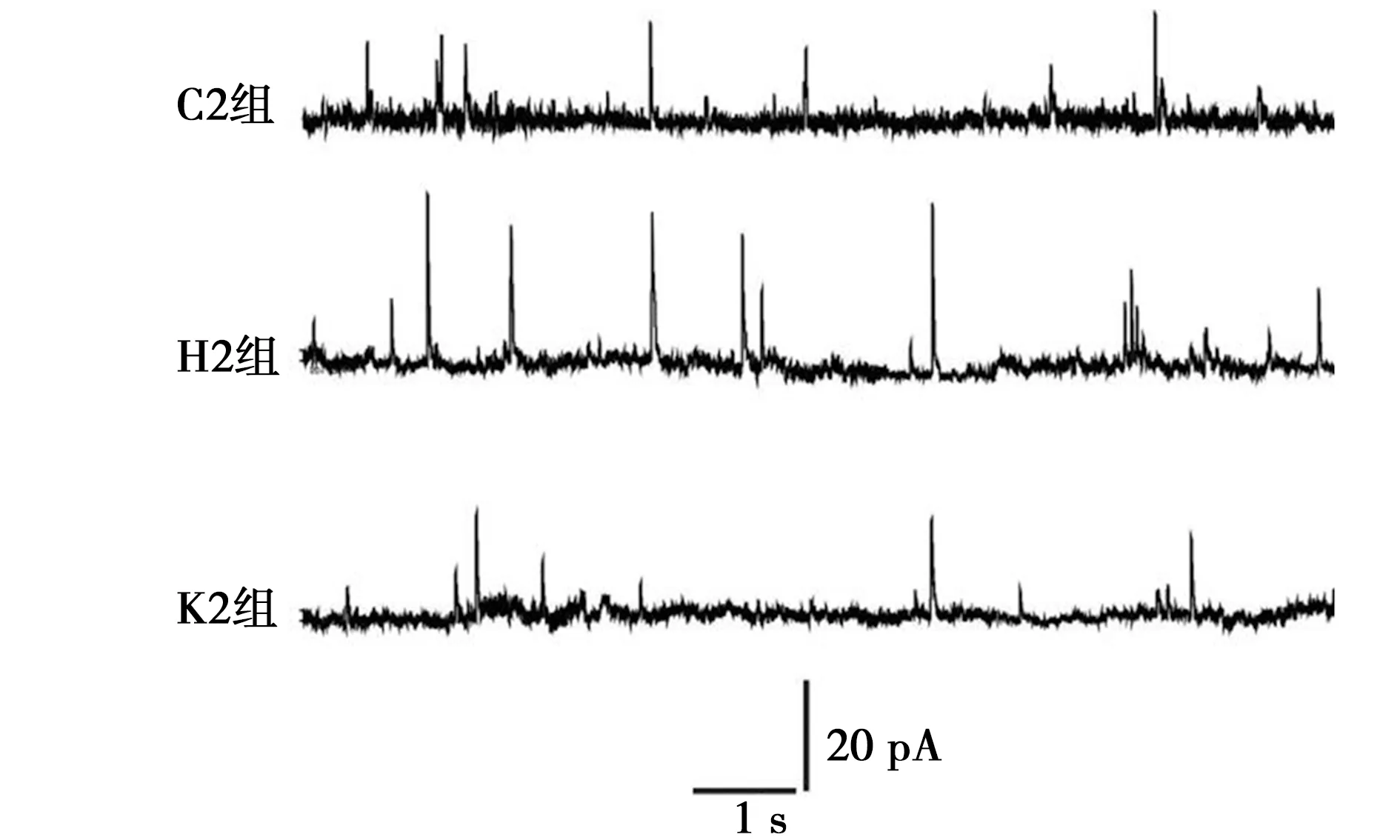
圖2 各組NMDAR mEPSCs的典型波形(10 s)
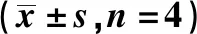
表2 各組mEPSCs的幅值和頻率比較
3 討 論
阿片藥物芬太尼主要用于臨床麻醉和鎮痛,但其有潛在的誘發OIH的風險,即采用阿片藥物鎮痛,卻反而引起機體對疼痛的敏感性增加〔9〕。這種矛盾的現象給臨床使用阿片類藥物帶來了困惑,亟待探明其機制,以指導臨床用藥和治療OIH。NMDAR廣泛存在于中樞神經系統,參與多種疼痛機制的調節〔10〕,包括神經病理性疼痛和中樞痛敏。已有研究表明,脊髓NMDAR參與阿片藥物瑞芬太尼誘導的OIH的產生〔11〕,但NMDAR是否參與脊髓上中樞神經系統來調節OIH尚待研究。本研究結果表明CeA NMDAR可能與芬太尼誘發的OIH產生有關。值得注意的是,NMDAR的拮抗劑氯胺酮,除了具有分離麻醉的特性外,還具有鎮痛、抗炎和抗抑郁的作用。不同劑量的氯胺酮所產生的作用不同,研究表明,氯胺酮產生麻醉作用所需的劑量遠大于其產生鎮痛、抗炎和抗抑郁作用所需的劑量〔12〕。鑒于氯胺酮的鎮痛作用與給藥劑量有關,本研究OIH大鼠單次腹腔注射低劑量(15 mg/kg)氯胺酮,該劑量遠低于產生麻醉作用所需的氯胺酮。且氯胺酮小鼠腹腔注射后的半衰期是13~25 min〔13~15〕,在注射30 min后,大鼠并未出現麻醉相關表現,大鼠完全清醒,活動自如,推斷氯胺酮此時主要產生的是鎮痛作用。在Abelaira等〔16〕研究中,給予大鼠15 mg/kg單次劑量的氯胺酮30 min后,即開始進行大鼠強迫游泳的行為學測試,說明大鼠是完全清醒的,也與本文觀察的結果一致。該研究結果提示,NMDAR拮抗劑氯胺酮可以改善OIH大鼠的疼痛閾值,產生鎮痛作用,從而緩解OIH。
NR2B是NMDAR執行調節功能的重要亞基,其磷酸化水平與NMDAR介導產生的慢性疼痛密切相關〔17〕,在NR2B亞基激活Ca2+內流之后,特定的鳥嘌呤核苷酸交換因子(GEFs)會刺激ERK的磷酸化(pERK)。pERK隨后從神經元細胞質轉位到細胞核。在細胞核中,pERK刺激核糖體激酶2,導致133絲氨酸上的環磷腺苷效應元件結合蛋白(CREB)磷酸化(pCREB)。pCREB進一步調節含有CRE的某些基因與CREB結合位點結合。通過與c-fos,神經激肽/P物質受體,腦源性神經營養因子(BDNF),降鈣素基因相關肽(CGRP)和環氧化酶(COX)-2等結合,導致傷害性感受反應的產生。這些神經調節劑會引發更多的NR2B亞基的激活,導致中樞神經系統的可塑性改變,這條途徑的最終結果是OIH和異常性疼痛的產生。本研究發現,OIH時,NMDAR亞基P-NR2B表達增加,給予NMDAR拮抗劑氯胺酮后,P-NR2B表達降低,可以逆轉OIH。
已有研究表明,NR2B亞基在NMDAR調控神經元突觸可塑性中起關鍵性作用,其表達水平與NMDAR產生的興奮性突觸傳遞正相關〔18〕。本研究中,OIH大鼠CeA NMDAR mEPSCs的幅值和頻率增加,給予NMDAR拮抗劑氯胺酮后,可以減少OIH大鼠CeA NMDAR介導的mEPSCs的幅值和頻率。mEPSCs的幅值體現的是作用于突觸后膜神經遞質釋放量,而mEPSCs的頻率體現的是突觸前膜神經遞質釋放概率,該研究結果提示OIH大鼠CeA神經元間突觸傳遞功能增強,CeA神經元間可能發生了功能上的突觸可塑性,且這種改變與NMDAR有關。本研究結果證實了阿片藥物芬太尼可能通過某種途徑作用于大鼠CeA神經元上的NMDAR亞基NR2B,從而調控神經元間突觸傳遞,使CeA發生突觸可塑性,從而產生OIH。但NMDAR是通過何種途徑調控大鼠OIH的產生尚待進一步研究。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
