高吸油、少皮膜PVC 樹脂的開發與研究
2023-11-27 06:35:48趙長森張奕齡
中國氯堿 2023年10期
關鍵詞:實驗
趙長森,張奕齡,牛 強
(1.內蒙古自治區聚氯乙烯特種樹脂工程技術研究中心,內蒙古 鄂爾多斯 016064;2. 內蒙古鄂爾多斯電力冶金集團股份有限公司,內蒙古 鄂爾多斯 016064)
PVC 樹脂一般可分為通用型樹脂和高端特種樹脂[1],相較于國外,國內對特種樹脂的開發較晚。21 世紀初, 隨著國內經濟的持續增長,PVC 行業迎來了高速發展,出現產能過剩,同時也迎來了高端特種樹脂發展的黃金時期, 特種樹脂的開發成為業內重點研究課題。
CPVC 是由PVC 經氯化改性而制得的一種介于橡膠和塑料之間的新型高分子熱塑性彈性體材料[2],是一種重要的化學改性材料。當前,國內CPVC 質量差,樹脂氯化程度不高,相較于國外存在較大差距,究其原因主要是缺少一種適合氯化的PVC 原料。
氯氣的擴散速度是提高氯化程度的關鍵所在,減少樹脂皮膜覆蓋率、制備內部疏松多孔、顆粒均勻的PVC 可以有效提高氯氣在PVC 分子中的擴散速度[2]。因此,研發一種適用于氯化的PVC 樹脂是一個重要的研究方向。 本文通過5L 聚合釜對該種樹脂進行小試開發,探究滿足氯化專用高吸油、少皮膜的PVC 樹脂合成工藝條件。
1 實驗部分
1.1 實驗原料
VCM,質量分數99.99%,內蒙古鄂爾多斯電力冶金集團;過氧化二碳酸雙(2- 乙基己酯)(EHP),配制為50%乳液, 鄂爾多斯瀚博科技有限公司;過氧化新癸酸異丙苯酯(CNP),配制為50%乳液,鄂爾多斯瀚博科技有限公司; 聚乙烯醇(PVA),配制為質量分數3%溶液,日本合成化學公司;羥丙基甲基纖維素(HPMC)60RT50,配制為質量分數3%溶液,山東泰安瑞泰纖維素有限公司; 聚乙烯醇BR-45,云南正邦科技有限公司;span60,上海泰坦科技股份有限公司;巰基乙醇,上海泰坦科技股份有限公司;碳酸氫鈉(NaHCO3),質量分數為96%,上海泰坦科技股份有限公司;聚氯乙烯(SG-5 型),鄂爾多斯電力冶金集團化工事業部PVC 分公司; 去離子水,二次蒸餾水,自制。
1.2 聚合實驗
在不銹鋼釜中加入一定量的去離子水、緩沖劑、分散劑、助分散劑、引發劑。充氮氣試壓,排氣抽真空排氧,使用氮氣進行多洗置換排氧。加入VCM,在室溫下進行30 min 冷攪拌,升溫至預設溫度進行恒溫聚合,控制溫變在±0.2 ℃,反應過程中使用計量泵將巰基乙醇流加入釜, 當釜內壓力下降0.05 MPa 時,加入終止劑結束反應,冷卻后停止攪拌,靜置、卸釜、出料、過濾、離心、干燥,檢測。
1.3 樣品分析依據[3,4]
按GB/T 5761-2018《懸浮法通用型聚氯乙烯樹脂》檢測PVC 樹脂的黏數、表觀密度、增塑劑吸收量、老化白度、250 μm 篩余物質量分數、63 μm 篩余物質量分數。按照GB/T 2917.1-2002《以氯乙烯均聚和共聚物為主的共混物及制品在高溫時放出氯化氫和任何其他酸性產物的測定剛果紅法》 使用剛果紅試紙法測試PVC 樹脂熱穩定性。 采用丹東百特Beettersize2600 激光粒度分布儀測試樹脂粒徑分布。 采用哈普RM-200C 轉矩流變儀測試樹脂塑化性能。采用場發射掃描電子顯微鏡SEM 測試樹脂表面皮膜。
1.4 數據記錄
溫度壓力曲線見圖1, 實驗過程中釜內溫度與壓力穩定,聚合時間為300 min。
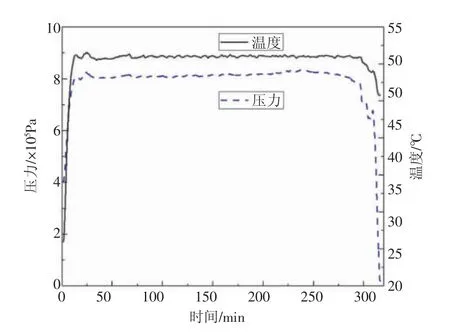
圖1 聚合釜內溫度、壓力曲線
2 結果與討論
2.1 聚合溫度的選擇
實驗預計生產聚合度趨于SG5 型PVC 樹脂,在正常生產過程中通用5 型樹脂生產溫度通常為57 ℃,考慮實驗設計中計劃使用鏈轉移劑巰基乙醇,鏈轉移劑的使用會降低PVC 的聚合度, 因此經過多次實驗確定了巰基乙醇的加入量,適當降低聚合溫度為51 ℃。
2.2 引發劑用量對增塑劑吸收率的影響
采用EHP 和CNP 兩種中高活性引發劑3∶1 復配,聚合溫度51 ℃,通過調整引發劑的用量研究其對增塑劑吸收量的影響,見圖2。
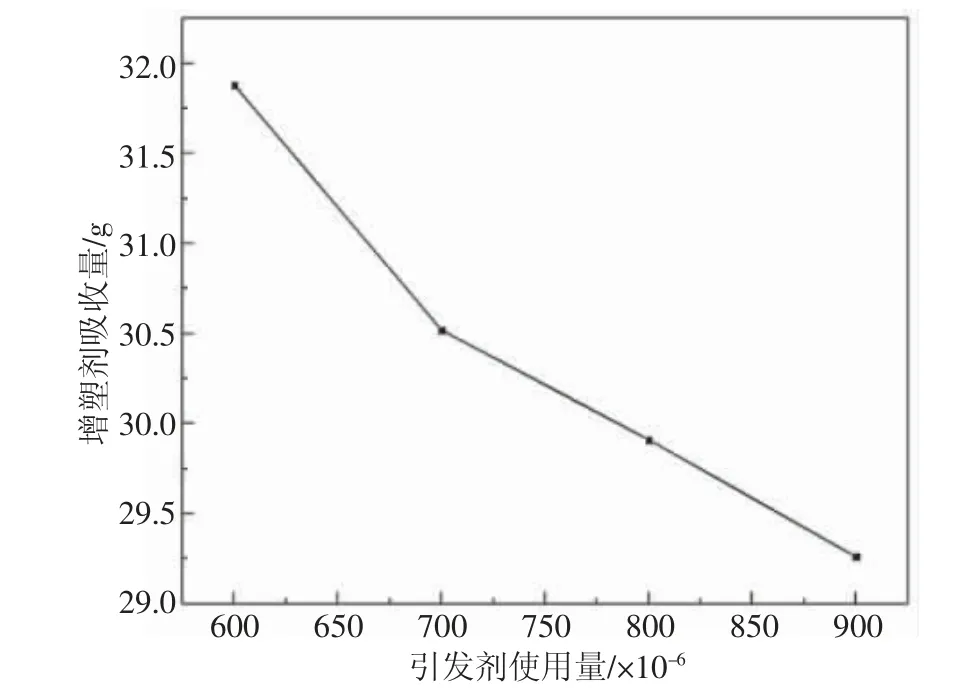
圖2 引發劑加入量對增塑劑吸收量的影響
隨著聚合過程中引發劑含量的增加, 增塑劑吸收量呈現遞減趨勢。 過量的引發劑會影響聚合過程初級粒子的數量和大小,加快聚合速度,導致單體液滴內形成的初級離子過度溶解,產生并聚,影響增塑劑吸收量。 因此,在聚合過程,在不影響實驗的基礎上,應控制引發劑的用量至最低[5]。
2.3 分散體系對樹脂性能的影響
分散劑和攪拌是影響懸浮聚合物顆粒度、 粒度分布、顆粒形態等特性的重要因素,在攪拌轉速不變的情況下,聚合實驗采用復合分散劑體系,分別使用強保膠能力PVA 和表面張力較低,分散效果優異的羥丙基甲基纖維為主要分散劑, 分別使用非離子表面活性劑Span60 和低醇解度的PVA 為助分散劑。
2.3.1 主分散劑對樹脂性能的影響
主分散劑采用HPMC 與W24N 復配,分別配制HPMC∶PVA 為1∶2 和3∶1 進行實驗, 在保持HPMC∶PVA=1∶2 比例不變的情況下, 提高主分散劑的用量進行實驗,實驗結果見表1 和表2。
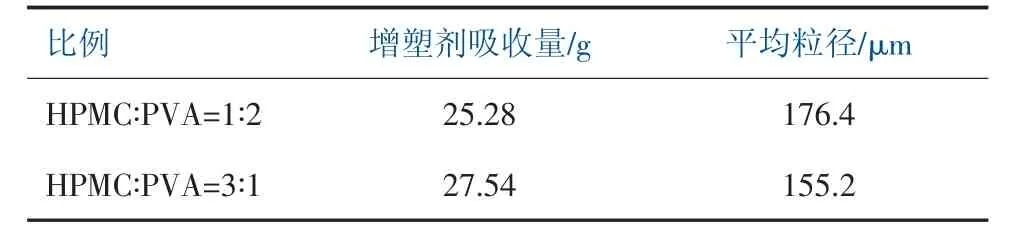
表1 調整主分散劑的比例對樹脂性能影響
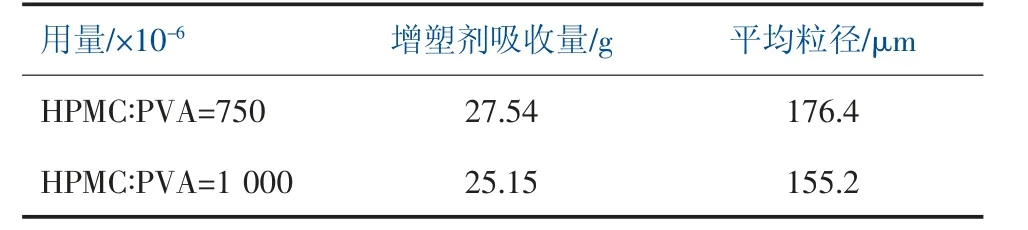
表2 調整主分散劑用量對樹脂性能影響
當聚合體系中,增大HPMC 的比例,增塑劑吸收量具有一定程度的提高,樹脂平均粒徑減小。在保持HPMC∶PVA=1∶2 比例不變的情況下,提高體系中分散劑的用量,增塑劑吸收量一定程度下降,樹脂平均粒徑減小。 由于采用高醇解度聚乙烯醇得到的PVC 樹脂外形利于提高PVC 樹脂的表觀密度,內部初級粒子凝并度高,孔隙率很小。隨著聚乙烯醇的減少,初級粒子的凝并度逐漸減少,孔隙率逐漸增加。而羥丙基甲基纖維素表面張力較低, 可以制得性能較好的疏松型樹脂,其樹脂顆粒疏松、增塑劑吸收量高[6]。HPMC 具有良好的分散效果,當體系中HPMC 的比例和用量增大,相應的樹脂的粒徑也會減小。 因此,在聚合分散體系中應盡量減少高醇解PVA 的用量。
2.3.2 助分散劑對增塑劑吸收量和平均粒徑的影響
在分散體系中加入油溶性好的表面活性劑,可以使分散劑主要分散在VCM 液滴表面, 可以加強對初級粒子的保護,穩定初級粒子,提高初級粒子尺寸,改善內部孔隙的均勻性,提高樹脂的增塑劑吸收量。 在實驗過程中, 分別使用非離子表面活性劑Span60 和低醇解度的PVA 為助分散劑進行實驗,結果見表3。
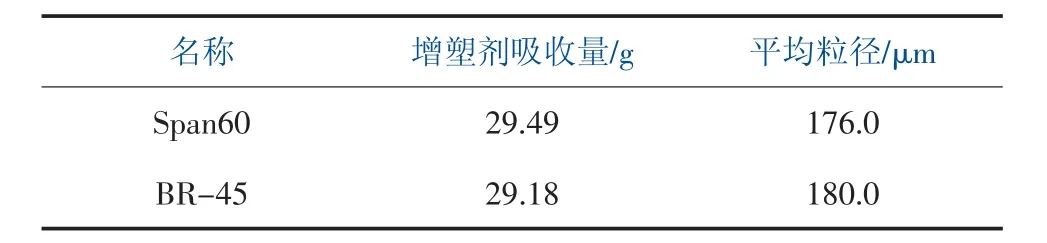
表3 不同助分散劑對樹脂性能的影響
通過表3 數據, 控制其他條件不變, 分別使用Span60 和BR-45 做助分散劑,對樹脂增塑吸收率都具有明顯提高,Span60 在增塑劑吸收量略高于BR-45,且粒徑更細,但二者效果相差不大,經過綜合考慮,選用Span60 為助分散劑。 調整助分散劑的比例和用量對樹脂性能影響見表4。
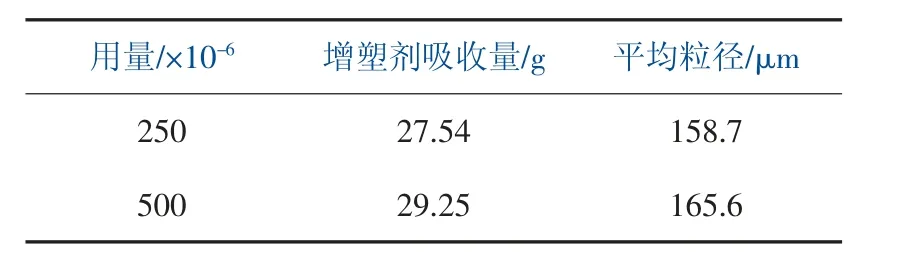
表4 調整助分散劑的比例和用量對樹脂性能影響
通過表4 中數據可得,控制其他條件不變,提高助分散劑的用量,樹脂增塑劑吸收量提高,樹脂平均粒徑變粗。這是由于非離子表面活性劑Span60 具有較強的油溶性,能夠進入油相液滴當中,隨著反應的進行及初級粒子的產生,Span60 會吸附在初級粒子表面,降低了固液界面張力,同時在初級粒子表面形成一層溶劑化膜, 使得初級粒子能夠穩定地存在于VC 液滴當中,降低聚集程度,提高了樹脂內部的孔隙率。由于助分散劑分子量相較于主分散劑更低,在液滴內難以形成緊密排列的分子, 能夠使主分散劑的膜強度減弱, 且油滴越小越容易發生合并,PVC的粒徑會增大。 在生產中可以在一定程度提高Span60 的用量,有利于樹脂增塑劑的吸收,但注意控制粒徑在合適范圍之內。
2.4 巰基乙醇對樹脂性能的影響
實驗過程中, 巰基乙醇作為鏈轉移劑參與到聚合過程當中,一方面調節聚合物的相對分子質量,控制聚合度, 使得在聚合溫度為51 ℃下生產的SG-3型樹脂轉變為SG-5 型樹脂, 另一方面具有提高增塑劑吸收量的作用。 分別通過探討巰基乙醇在聚合中的加入量和加入方式兩個方面對PVC 樹脂顆粒性能的影響。
2.4.1 巰基乙醇的加入量對樹脂性能的影響
在聚合過程中分別加入100×10-6、200×10-6、300×10-6、400×10-6巰基乙醇, 加入方式為反應開始90 min 后滴加的方式入釜, 控制90 min 內滴加完畢。 巰基乙醇的加入量對樹脂性能影響見表5。
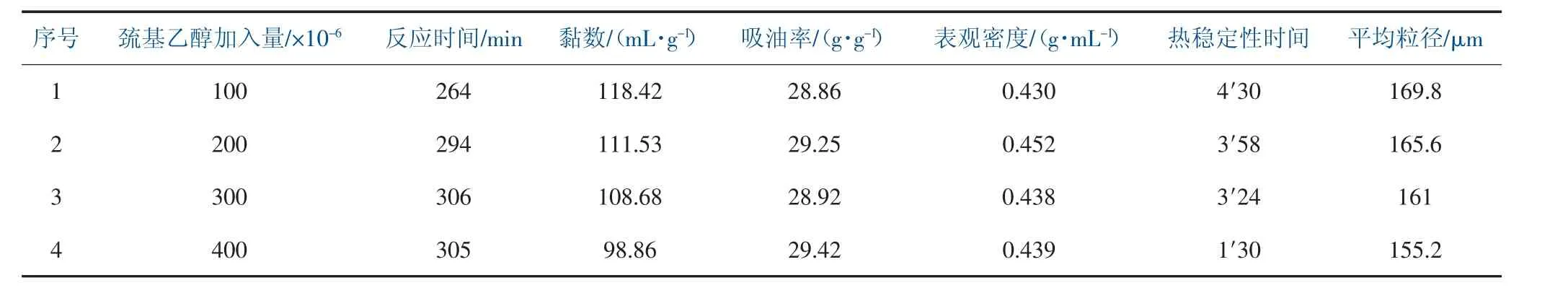
表5 巰基乙醇的加入量對樹脂性能影響
通過表5 中數據可得, 隨著巰基乙醇的含量增加,相應的黏數有了明顯的遞減趨勢,進而控制了樹脂的平均聚合度;反應時間先上升后趨于平緩,由于巰基乙醇是一種還原性物質,在聚合過程中,遇到過氧化性的引發劑會進一步消耗引發劑, 因此實際參與聚合反應引發劑減少, 在實驗過程中需相應提高引發劑的用量,一定程度控制聚合反應速度和反應時間;吸油率微量增加,實驗結果表明巰基乙醇并未對增塑劑吸收量有較大影響, 可能由于本文實驗方案通過多種方式提高增塑劑吸收量, 樹脂吸油量達到30 g 左右,趨于一定飽和狀態,且實驗中使用的巰基乙醇含量較少,不能觀測到明顯的變化;隨著巰基乙醇的含量增加,熱穩定性時間逐步降低,在實驗體系中,巰基乙醇會影響分散劑體系,破壞其保膠能力,而分散劑對熱穩定性能在一定程度上產生影響,因此在聚合實驗中,應盡量控制巰基乙醇的含量,最好不超過300×10-6;隨著巰基乙醇的含量增加,樹脂的平均粒徑變小,但變化幅度不大。 由于巰基乙醇在低濃度范圍內, 具有降低表面活性的作用,巰基乙醇的用量增加,分散體系的分散能力提高,保膠能力下降[8]。
2.4.2 巰基乙醇的加入方式對樹脂性能的影響
聚合實驗中,采用巰基乙醇分兩次加入的方式,將巰基乙醇溶于一定量水中,反應前期加入一部分,其余量在反應開始90 min 后通過計量泵滴加入釜,控制滴加速度和時間, 研究不同加入方式對樹脂顆粒性能的影響, 巰基乙醇加入方式對樹脂性能影響見表6。
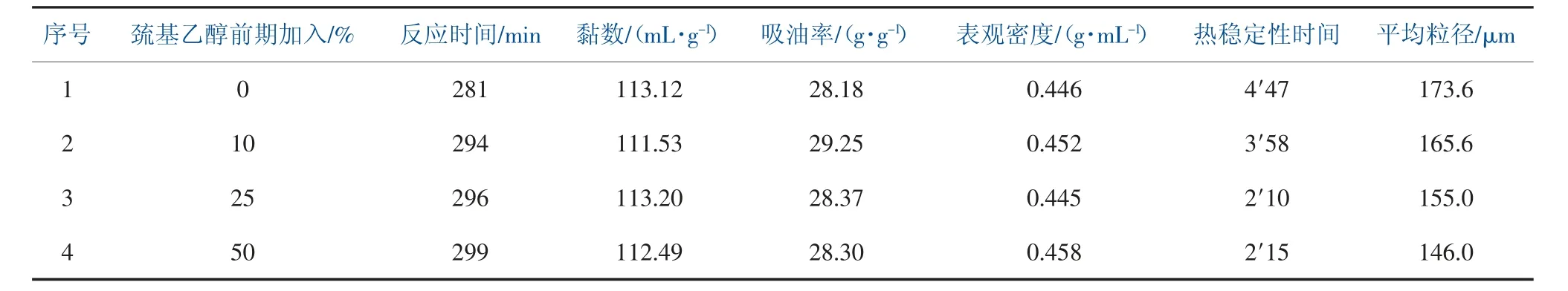
表6 巰基乙醇加入方式對樹脂性能影響
根據表6 中數據可得, 隨著第一次加入巰基乙醇的增加, 聚合反應的時間有了一定程度的延長,樹脂聚合度變化不大,吸油率先上升后下降,表觀密度變化不大,熱穩定性能逐漸下降,樹脂平均粒徑減小。因此,前期不加巰基乙醇,樹脂熱穩定性更好,前期加入10%巰基乙醇,粒徑更小,吸油率更高。
2.5 緩沖劑的選擇
在聚合過程中,實驗中常用NaHCO3、NH4HCO3、NaOH 等作為緩沖劑調節體系pH。 根據史悠彰等人介紹[7],在體系中添加界面阻聚劑能夠阻止PVC 大分子自由基與分散劑保護膠發生接枝共聚反應,從而改善樹脂顆粒皮膜結構。因此使用NH4HCO3作為阻聚劑既可以調節體系pH, 也可以改善樹脂的皮膜,提高樹脂增塑劑吸收量。
2.6 樹脂皮膜SEM 測試
樹脂的皮膜主要是分散劑接枝共聚包覆在粒子表面,因此可以通過調節分散體系,減少分散劑的用量、選用合適的助分散劑、阻聚劑NH4HCO3、添加巰基乙醇等方式破壞樹脂皮膜,減少皮膜包覆率。通過掃描電鏡測試的PVC 表面皮膜狀態見圖3。

圖3 引發劑的加入量對增塑劑吸收量的影響
由圖3 可以看出,部分樹脂表面沒有皮膜,部分樹脂皮膜遭到破壞,內部疏松多孔,更易于增塑劑的吸收,能夠一定程度加快氯氣的擴散速度,樹脂粒徑略微大于100 μm。樹脂的皮膜可能由于實驗中減少PVA 的用量,采用低醇解度PVA 和羥丙基甲基纖維素作為主要分散體系。 巰基乙醇能夠一定程度醇解PVA, 進而破壞包覆于粒子表面的皮膜。 助分散劑Span60 的加入也一定程度改善了PVC 樹脂的皮膜形態,使得樹脂內破疏松多孔,是由于Span60 優先于VC-H2O 界面吸附, 進而破壞了界面上形成的樹脂皮膜[8]。
2.7 塑化性能測試
使用哈普RM-200C 轉矩流變儀對生產的實驗樹脂和SG-5 型通用樹脂進行塑化性能測試, 測試條件為測試溫度180 ℃,轉速為35 r/min,配鈣鋅穩定劑4 份/100 g,樹脂填充量52 g,實驗制備樹脂與SG-5 型樹脂塑化扭矩與時間分布圖,見圖4。
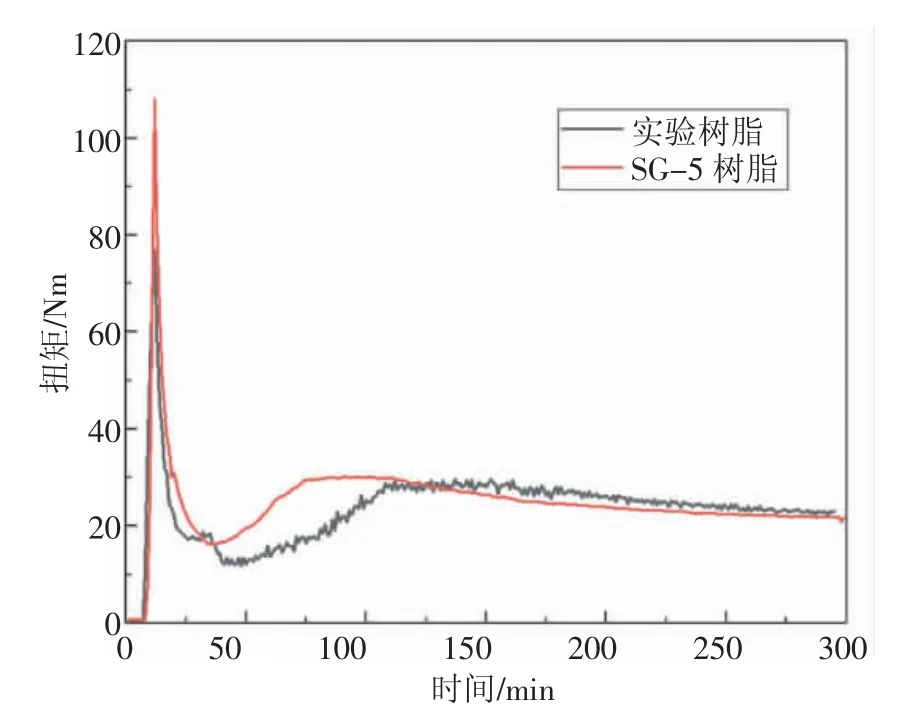
圖4 實驗制備樹脂與SG-5型樹脂塑化扭矩與時間分布圖
從圖4 可以看出, 相較于SG-5 型樹脂的塑化曲線, 實驗制備樹脂在位于裝載峰與最小扭矩處出現明顯的塑化拐點, 表明原料中可能含有兩種不同特性的樹脂, 由此猜想可能由于該樹脂皮膜覆蓋存在不同形式,部分樹脂皮膜較為完整,部分樹脂皮膜被破壞,導致出現兩種塑化速度,部分樹脂優先于其他樹脂塑化,與SEM 結果相符合。
2.8 樹脂粒徑分布測試
實驗制得型樹脂的粒徑分布圖見圖5,10%的樹脂粒徑小于90.64 μm,50%的樹脂粒徑小于160.5 μm,90%的樹脂粒徑小于248.2 μm, 其比表面積平均粒徑為139.6 μm,體積平均粒徑為165.6 μm。 圖5 還列出來生產線SG-5 樹脂粒徑分布,可以得出實驗樹脂粒徑跨度略大于SG-5 型樹脂, 粒徑大小優于SG-5 型樹脂。 由于生產等溫入料,不同于實驗存在升溫過程,樹脂粒徑分布更加平均,樹脂跨度更小。
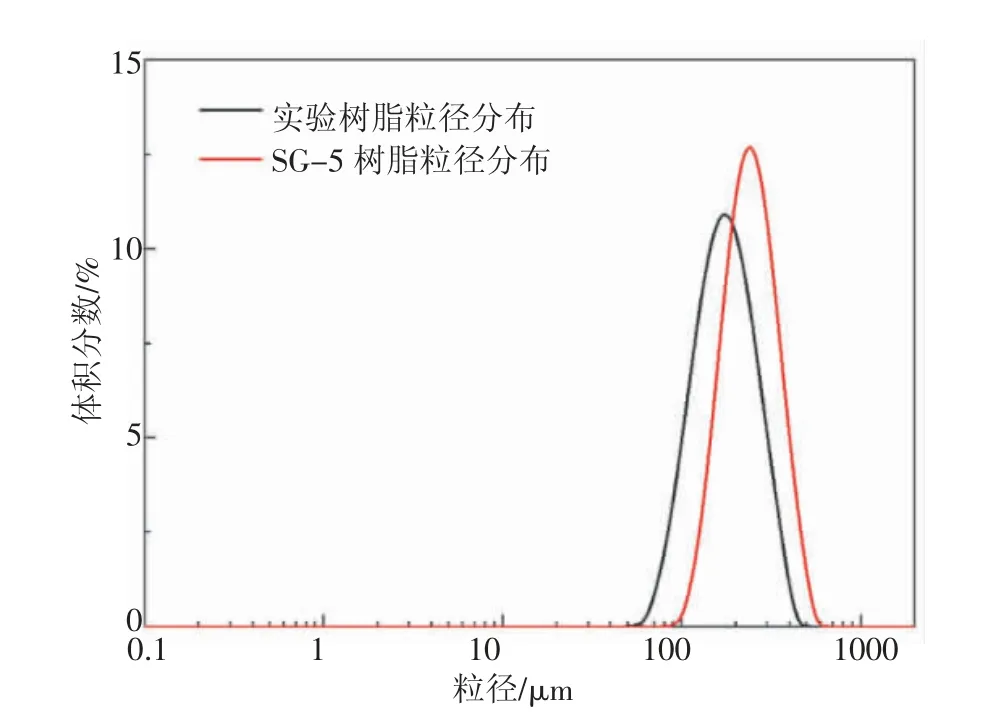
圖5 實驗樹脂與SG-5型樹脂粒徑分布
3 結論
本文通過優化工藝條件, 制備了一種增塑劑吸收量高, 皮膜覆蓋率低適用于氯化制CPVC 的PVC樹脂原料。 測定了聚合分散體系、緩沖劑、鏈轉移劑的加入量及加入方式等對PVC 樹脂性能的影響,得到結論如下。
(1)通過減少主分散劑中PVA 的用量,在分散體系中加入表面活性劑Span60 有利于樹脂增塑劑的吸收。
(2)NH4HCO3既可以作為緩沖劑調節聚合體系的pH,又能夠充當阻聚劑,提高樹脂增塑劑吸油量,改善樹脂皮膜覆蓋率。
(3)通過調節巰基乙醇控制其合理的加入方式和加入量,能夠有效提高樹脂增塑劑吸收量,控制樹脂的粒徑大小。
(4)可以通過控制主分散劑的種類,添加助分散劑、阻聚劑等幾種方式改善樹脂的皮膜。
本次實驗通過5L 聚合小釜制備的該種樹脂增塑劑吸收量高、樹脂內部疏松多孔、皮膜覆蓋率低,能夠有效提高氯氣在PVC 分子中的擴散速度,適用于氯化CPVC 的原料。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
