液相色譜-氫化物發生-原子熒光光譜法測定中藥樣品中的4種砷形態
2024-01-16 03:19:14劉德曄張琳昀
食品與藥品 2023年6期
張 雯,徐 麗,劉德曄,張琳昀*
(1.江蘇省疾病預防控制中心,江蘇 南京 210009;2.駐馬店市食品藥品檢驗所,河南 駐馬店 463000)
中藥材中的重金屬及有害元素的污染一直是國內外普遍關心的問題[1]。隨著檢測技術的進步,僅測定中藥材中的重金屬總量已不能滿足監測其毒性的要求。同一元素的不同化學形態其毒性存在很大差異[2]。砷是一種廣泛存在于自然界中的重金屬[3-4],砷對人體的毒性不僅與其總量有關,更與其存在形態有關。砷存在的主要形態可分為有機砷和無機砷兩大類,無機砷主要包括砷酸鹽[As(V)]和亞砷酸鹽[As(III)],有機砷包括一甲基砷(MMA)和二甲基砷(DMA)等,無機砷的毒性比有機砷的毒性強,而在海洋生物中富集較多的有機砷如砷膽堿(AsC)和砷甜菜堿(AsB)等毒性相對較小[5]。研究發現,砷各種形態的毒性大小依次為As(III)>As(V)>MMA>DMA>AsB。無機砷化合物已被國際癌癥研究機構確認為I類致癌物[6]。無機砷攝入過量易導致皮膚、神經、消化、生殖、免疫系統等損傷,更加嚴重的會導致肝癌、皮膚癌等癌癥的發生[7]。2020年版《中華人民共和國藥典(一部)》規定了 28種中藥材及6個中成藥中重金屬及有害元素限量,《藥用植物及制劑 進出口綠色行業標準》規定中藥重金屬的一般限量標準為砷≤2.0 mg/kg[8-9]。考慮到無機砷的高毒性,中藥中砷的限量標準顯然不夠完善。
常用的砷形態的測定方法主要是食品安全國家標準GB5009.11-2014[10]中的第一法液相色譜-原子熒光法(LC-AFS)[11-12]和第二法高效液相色譜-電感耦合等離子體質譜法(LC-ICP-MS)[13-15],以上兩種方法亦可作為中藥材中無機砷測定方法。除此之外還有高效液相色譜-電感耦合等離子體串聯質譜法(HPLC-ICP-MS/MS)[16-17],但由于儀器昂貴,運行成本較高,難以被普及。本研究在國標第一法LC-AFS方法上進行改進,優化了前處理條件,采用Princen砷形態快速分析柱,選擇合適的試劑條件和儀器參數,建立一種能同時測定中藥材樣品中As(III)、As(V)、DMA和MMA 4種砷形態的方法,對完善中藥材中無機砷的限量標準,正確評估土壤環境中砷的污染程度有重要價值,也可為食品和環境監管提供數據支持。
1 儀器與試藥
1.1 儀器
DHG-9076A型電熱恒溫鼓風干燥箱(上海精宏);DEENA3型全自動石墨消解儀(上海儀真);3-18ks型高速離心機(德國Sigma公司);E180H型超聲波清洗機(德國Elma公司);Elspe-2型液相色譜儀(廣州譜臨晟);BAF-4000型原子熒光光譜儀(北京寶德)。
1.2 試藥
砷酸根[A s(V)]標準溶液(編號:GBW08667,17.5 μg/g),亞砷酸根[As(III)]標準溶液(編號:GBW08666,75.7 μg/g),MMA標準溶液(編號:GBW08668,25.1 μg/g),DMA標準溶液(編號:GBW08669,52.9 μg/g,中國計量科學研究院);鹽酸,硝酸(優級純,南京化學試劑股份有限公司);磷酸氫二鉀(分析純,南京化學試劑股份有限公司);氫氧化鉀,氨水,硝酸銨(分析純,國藥集團);硼氫化鉀(分析純,旭日成化學);高純氬氣(純度≥99.99 %,南京文達特種氣體有限公司);0.45 μm水系濾膜;50 ml聚丙烯離心管。實驗用水均為超純水。本研究所用的中藥材均為河南省駐馬店市市場監督管理局抽檢樣品。
2 方法與結果
2.1 LC-AFS條件
2.1.1 LC條件 色譜柱:Princen砷形態快速分析柱(As Spec Fast Analysis Column,4.6 mm×50.0 mm,5.0 μm);保護柱:Princen保護柱(As Spec Guard Column,4.6 mm×50.0 mm,10.0 μm);載氣:氬氣,壓力:0.15 MPa。流動相:流動相A:5 mmol/L 磷酸氫二鉀+1 mmol/L硝酸銨,氨水調節pH 10.9;流動相B:25 mmol/L磷酸氫二鉀+40 mmol/L硝酸銨,氨水調節pH 9.2; 梯度洗脫程序:0~102 s,100 %流動相A;103~241 s,100 %流動相B;242~360 s,100 %流動相A。流速:1.2 ml/min;進樣量:100 μl。
2.1.2 AFS檢測條件 載流:5 %鹽酸溶液;還原劑:0.5 %氫氧化鉀溶液+1.5 %硼氫化鉀溶液;負高壓:290 V;燈電流:60 mA;輔助電流:60 mA;載氣流速:400 ml/min;輔助氣流速:800 ml/min。氫氣發生器流量:100 ml/min。
2.2 4種不同形態砷含量測定法
2.2.1 標準溶液的配制 準確稱取亞砷酸根[As(III)]1.3210 g、砷酸根[As(V)]5.7150 g、MMA 3.9880 g、DMA標準溶液1.8904 g,分別置于10 ml量瓶中,加水定容。所得溶液中As(III)、As(V)、MMA、DMA濃度均為10 μg/ml。分別準確吸取各形態砷標準溶液(10 μg/ml)各1 ml于10 ml量瓶中,加純水定容,得1.0 μg/ml混合標準溶液(以不同形態的砷計)。
2.2.2 不同形態砷含量測定 稱取烘干粉碎后的中藥試樣0.5 g,置于50 ml消解罐中,加入0.15 mol/L硝酸溶液20 ml,浸泡過夜,經全自動石墨消解儀90 ℃熱浸提2.5 h,每0.5 h振搖1 min。提取完畢后,提取液冷卻至室溫,8000 r/min離心10 min,吸取上層清液,0.45 μm濾膜過濾后上機測定。同時按同一操作方法制備空白溶液。按2.1.1項下色譜條件進樣,按2.1.2項下條件檢測。
2.3 總砷測定
參考GB 5009.11-2014《食品安全國家標準 食品中總砷及無機砷的測定》中的原子熒光光譜法,稱取烘干粉碎后的中藥試樣1.0 g,置于坩堝中,加入硝酸鎂溶液,混勻,再加入1 g氧化鎂覆蓋樣品,于電爐上炭化至無黑煙后,移入550 ℃馬弗爐灰化4 h,待樣品冷卻后加入鹽酸溶液中和氧化鎂并溶解灰分,轉入25 ml量瓶中,加入2 ml硫脲-抗壞血酸溶液后定容至刻度,混勻放置后,按2.1.2項下條件測定總砷含量。
2.4 檢測條件的優化
2.4.1 前處理方法的優化 GB 5009.11-2014液相色譜-原子熒光光光譜法[10]測定無機砷中,樣品提取方式為90 ℃熱浸提2.5 h,每0.5 h振搖1 min,需在干燥箱加熱,手動振搖。采用全自動石墨消解儀,可自動設置程序完成升溫、每0.5 h振搖1 min以及降溫的整個過程,試驗結束后自動冷卻,自帶的特氟龍消解罐能四周加熱,保證受熱均勻,且無需人為操作。以當歸為對象,對比以往文獻中使用較多的60 ℃超聲加熱1 h[16]、90 ℃干燥箱熱浸提2.5 h及90 ℃全自動石墨消解儀提取2.5 h 3種提取方式,計算3種方式下提取出的砷的總量,結果見圖1。結果表明,60 ℃超聲加熱1 h提取效率最低,90 ℃干燥箱熱浸提及全自動石墨消解儀提取效果相當,由于全自動石墨消解儀操作方便,綜合考慮,選擇此種消解方式。
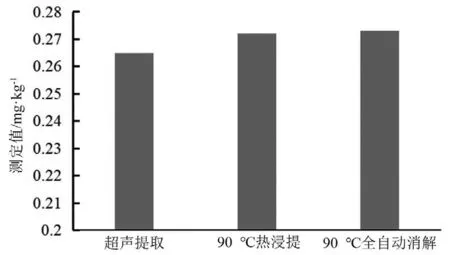
圖1 不同提取方式下的4種砷形態測定值
2.4.2 色譜柱的優化 試驗對比了兩種色譜柱,即Hamilton PRP-X100陰離子交換色譜柱(250 mm×4 mm)和Princen砷形態快速分析柱(150 mm×4 mm)對同一濃度的無機砷標準品的分析結果。結果顯示兩種色譜柱均能有效分離4種砷形態組分,但出峰時間差異較大。兩種色譜柱各組分的出峰時間對比見圖2。使用Hamilton PRP-X100陰離子交換色譜柱,4種砷形態出峰時間需近1200 s,分析時間較長,而使用Princen砷形態快速分析柱可將分析總時間控制在360 s內,可見Princen砷形態快速分析柱分析速度快,檢測效率高,且各組分分離良好。

圖2 兩種色譜柱各組分出峰時間對比
2.4.3 流動相的選擇 處理后樣品中的砷是以陰離子形態存在的,實驗應選擇陰離子交換色譜柱進行分離。基于國標方法,選擇磷酸鹽溶液作為流動相,流動相的pH值是決定分離效果的影響因素之一。由于經過前處理后的樣品溶液呈酸性,進入色譜柱會影響樣品的保留能力和分離度,為了中和樣品的酸性,選擇堿性的流動相可達到更好的分離效果,同時可延長色譜柱壽命。結果顯示,流動相的堿性較弱不利于峰的分離,若堿性過強(加入氨水調節pH值),則本底值上升。綜合考慮,確定流動相A:5 mmol/L磷酸氫二鉀+1 mmol/L硝酸銨,pH 10.9;流動相B:25 mmol/L磷酸氫二鉀+40 mmol/L硝酸銨,pH 9.2,采用梯度洗脫,保證良好的分離效果。
2.4.4 AFS檢測條件的優化
2.4.4.1 載流的濃度 實驗過程中,載流與硼氫化鉀反應生成氫氣,將三價砷還原成氣態氫化物,一般選擇鹽酸溶液。以20 μg/L混合標準溶液為考察對象,分別考察了3 %,4 %,5 %,6 %,7 %鹽酸溶液作為載流對待測物峰面積的影響,結果見圖3。鹽酸濃度為3 %時,4種待測物峰面積明顯偏低,隨著濃度增加,峰面積逐步上升,后趨于平衡,以6 %,7 %鹽酸為載流時的峰面積與以5 %鹽酸為載流時基本保持一致,證明5 %鹽酸溶液即可滿足要求,故實驗選擇5 %鹽酸溶液為載流。

圖3 鹽酸濃度對4種砷形態峰面積的影響
2.4.4.2 硼氫化鉀的濃度 確定用5 %鹽酸溶液作為載流后,以20 μg/L混合標準溶液為考察對象,分別考察了氫氧化鉀溶液濃度為0.5 %,以1 %,1.5 %,2 %,2.5 %,3 %硼氫化鉀溶液作為還原劑對峰面積的影響,結果見圖4。由圖4可見,隨著硼氫化鉀濃度的升高,4種砷形態的峰面積明顯增大,當濃度達1.5 %后,峰面積趨于平穩,說明1.5 %硼氫化鉀溶液即可滿足要求,故選擇1.5 %硼氫化鉀溶液為還原劑。

圖4 硼氫化鉀溶液濃度對4種砷形態峰面積的影響
2.5 方法學考察
2.5.1 專屬性實驗 分別吸取2.2.1項下4種砷形態混合標準溶液(1.0 μg/ml)及2.2.2項下陰性樣品溶液(空白溶液)、當歸樣品溶液各100 μl,按2.2項下方法進樣測定,色譜圖見圖5。結果表明,4種砷形態成分均達到基線分離,與其他成分間均無明顯干擾,表明檢測方法的專屬性良好。

圖5 專屬性實驗圖譜
2.5.2 線性范圍及相關系數 精確吸取一定體積的混合標準溶液,分別用0.15 mol/L硝酸溶液配制濃度為0,1.0,2.0,5.0,10.0,20.0,50.0 μg/L的系列標準溶液,按2.2項下方法檢測,記錄色譜圖。分別以4種砷形態的濃度(μg/L)為橫坐標,以其對應的峰面積為縱坐標,繪制標準曲線。方法的線性方程和相關系數見表1。在選定的色譜條件下,標準曲線在1~50 μg/L范圍內線性良好,相關系數(r)均>0.999。
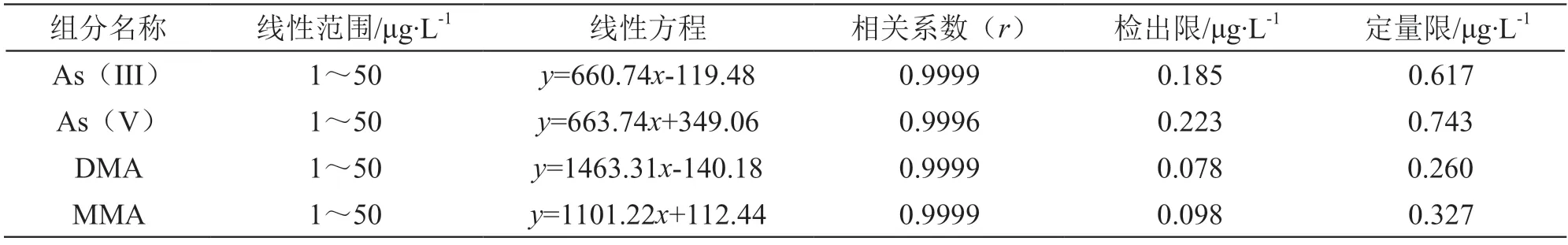
表1 方法的線性范圍、線性方程、檢出限及定量限
2.5.3 檢出限和定量限 采用逐級稀釋的方式,以3倍基線噪聲時砷形態的濃度為檢出限(S/N=3),以10倍基線噪聲時砷形態的濃度為定量限,結果見表1。實驗中如稱取固體樣品1.0 g,定容至20 ml時,As(III)、As(V)、DMA和MMA的檢出限依次為0.00370,0.00446,0.00156,0.00196 mg/kg,定量限依次為0.0123,0.0149,0.00520,0.00654 mg/kg。完全可滿足中藥樣品中無機砷的分析要求。
2.5.4 精密度和回收率試驗 精密稱取已測得砷含量的當歸樣品0.5 g,分別準確加入低、中、高3個濃度水平的4種砷形態混合標準溶液,根據已優化的實驗條件對加標樣品做回收率試驗,加標回收率范圍為96.7 %~101.9 %。每個濃度制備6個平行樣品,進行精密度試驗,計算相對標準偏差(RSD),結果見表2。結果表明方法的準確度和精密度均符合檢測要求。

表2 當歸樣品回收率及RSD值
2.5.5 穩定性實驗 取同一批當歸樣品,加入一定濃度的砷混合標準溶液,按2.2.2項下方法處理,置于4 ℃條件下,分別于0,2,4,6,8,12 h,按2.1.1項下色譜條件進樣,按2.1.2項下條件檢測。經測算,As(III)、As(V)、DMA、MMA峰面積RSD依次為2.1 %,1.9 %,0.9 %,1.6 %,表明樣品溶液在4 ℃條件下放置12 h基本穩定。
2.6 實際樣品分析
采用本方法測定市售的7種28批次的中藥材中的4種砷形態,同時測定其總砷含量,計算其無機砷含量與總砷含量的百分比。結果見表3。
由表3可見,各批次樣品均未檢出MMA,僅有黃芪和黃芩中的2批次檢出少量DMA,除了甘草1批次未檢出As(III)外,其余批次全部檢出As(III)和As(V),說明本次檢測的中藥材中砷形態主要以As(III)和As(V)存在,且As(V)含量均高于As(III),說明中藥樣品中的無機砷多以As(V)形式存在。2020年版《中華人民共和國藥典(一部)》[8]對中藥材的重金屬限量要求為砷≤2.0 mg/kg,通過檢測中藥材中的總砷含量,可看出此次檢測的中藥材中總砷均未超標,無機砷總量占總砷總量的比例為14 %~93 %,表明各樣品中不僅存在不同比例的As(III)和As(V),且有未能檢出的其他有機砷形態,但由于無機砷的危害遠大于有機砷,所以測定樣品中的無機砷即可表明中藥材的食用風險。
3 結論
本文采用液相色譜-氫化物發生-原子熒光光譜法測定中藥材中的4種砷形態。與GB5009.11-2014國標方法相比較,優化了前處理條件,選擇了操作更加簡便的全自動石墨消解儀,簡化了實驗操作步驟。同時優選出峰時間更快的Princen砷形態快速分析柱分離樣品,使樣品在360 s內完全分離,極大地縮短了樣品檢測時間,節約了試劑和人工成本。本方法檢出限低、靈敏度高,各待測組分回收率為96.7 %~101.9 %,RSD均小于3 %,說明方法重復性好,準確度和精密度均能滿足方法學要求。綜上,本方法在定量分析中藥材中As(III)、As(V)、DMA和MMA 4種砷形態中可取得令人滿意的結果,值得普及推廣。
