噬血細胞綜合征1例報道并文獻復習
2024-01-18 01:41:00藍方權洪峰周璇
右江醫學 2023年12期
藍方權,洪峰,周璇
(1.安徽中醫藥大學研究生院,安徽合肥230012;2.安徽中醫藥大學第一附屬醫院綜合ICU,安徽合肥230011)
噬血細胞綜合征(hemophagocytic syndrome, HPS)又叫噬血細胞性淋巴組織細胞增生癥(hemophagocytic lymphohistiocytosis, HLH),是一種由淋巴細胞和巨噬細胞過度活化,損及多個臟器甚至危及生命的過度炎癥反應綜合征,即使規范治療,死亡率仍極高[1]。本病病情發展迅速,可由多種病因引起,其中感染相關性HPS癥狀與膿毒癥及全身炎癥反應綜合征具有相類似特征,導致診斷難度極大,易漏診誤診,因此及早診斷并適時治療對提高HPS患者存活率極為重要[2]。本文就我院綜合ICU于2022年4月23日收治的1例老年男性HPS病例進行報道如下。
1 病例介紹
1.1 既往資料患者,男,69歲,分別在2021年8月、2021年12月無明顯誘因下出現反復發熱,體溫最高達40 ℃,反復就診于外院。2022年3月10日再次出現發熱,于當地治療無效后轉至上級醫院治療,考慮肺部感染,治療后癥狀未見好轉。遂于2022年4月6日轉至另一家醫院治療,完善結核斑點試驗、結核Xpert、痰液結核菌培養等檢查,排除活動性肺結核后予相關處理,體溫控制仍欠佳。于2022年4月13日再次轉至第三家醫院,完善PET/CT及相關檢查未發現腫瘤病變(見圖1),全血宏基因組二代測序(mNGS)提示奧斯陸莫拉氏菌(序列數1),予抗感染等治療后,效果仍不佳,10天后患者及家屬要求出院。
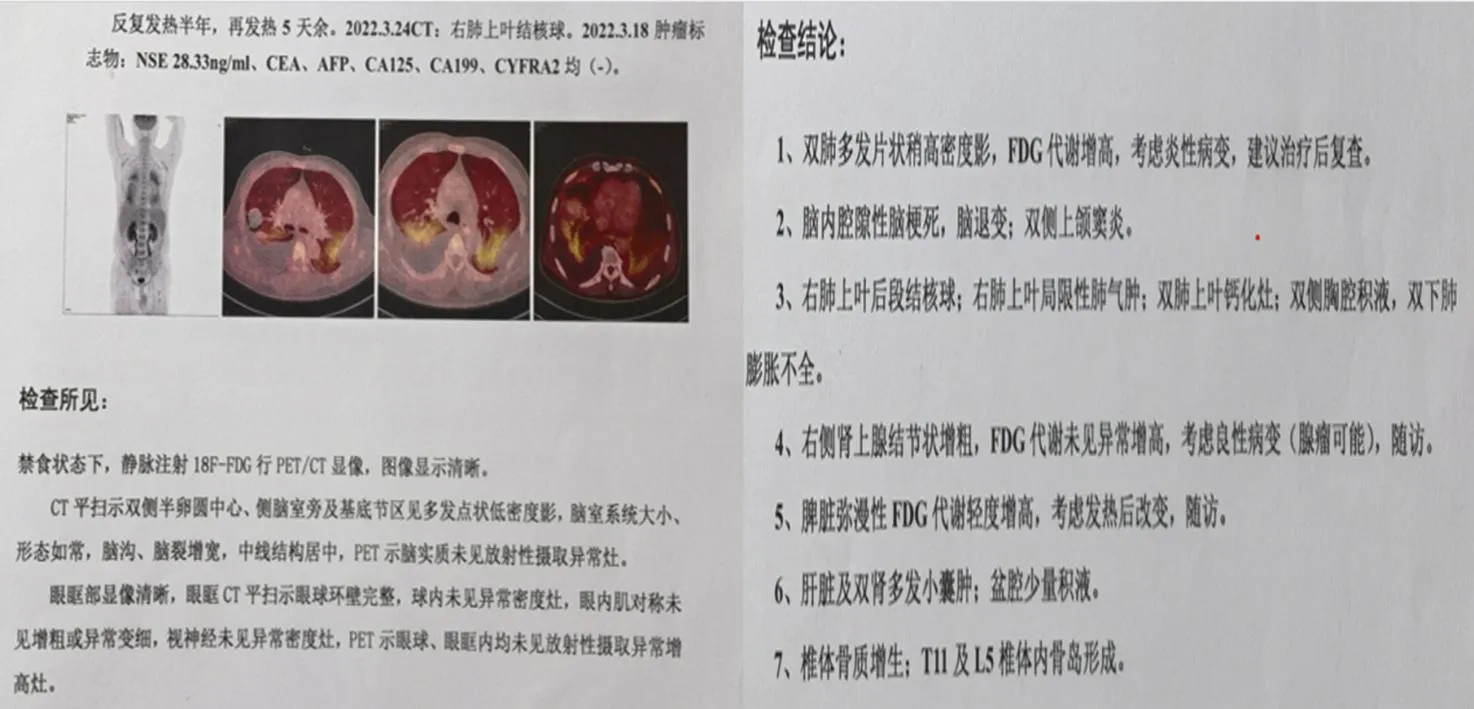
圖1 PET/CT檢查結果
1.2 本次入院檢查資料患者出院2小時后再次呼吸困難伴意識模糊、煩躁及惡心干嘔,醫療救護120接至我院急診科,心電監護提示指脈氧60%~70%波動,血壓不能維持,最低至80/40 mmHg(1 mmHg=0.133 kPa),心率達150次/分,給予氣管插管后接呼吸機輔助呼吸,排除新型冠狀病毒感染后,轉我科進一步診治。轉入查體:T 38.8 ℃,P 120次/min,R 25次/min,BP 110/87 mmHg,意識模糊,急病面容,查體不合作;雙瞳等圓,直徑約3 mm,光敏;雙肺呼吸音稍低,可聞及少許干濕性啰音;心率120次/分,律齊;雙下肢中度凹陷性水腫。輔助檢查:血氣分析(機械通氣后)示乳酸1.7 mmol/L,二氧化碳分壓30.3 mmHg,pH7.48,氧分壓117 mmHg;血常規示白細胞計數(WBC)2.77×109/L↓,血紅蛋白(HGB)70 g/L↓,血小板計數101×109/L,紅細胞計數2.51×1012/L↓;血生化示白蛋白25 g/L↓,降鈣素原(PCT)7.117 ng/mL,C反應蛋白95.74 mg/L↑,血清淀粉樣蛋白A152.76 mg/L↑;胸部CT示兩肺炎癥及陳舊灶,右肺局限性肺氣腫;雙側肺氣腫伴節段性肺不張;心包積液(見圖2)。初步診斷:(1)重癥肺炎;(2)膿毒性休克;(3)低蛋白血癥;(4)中度貧血。
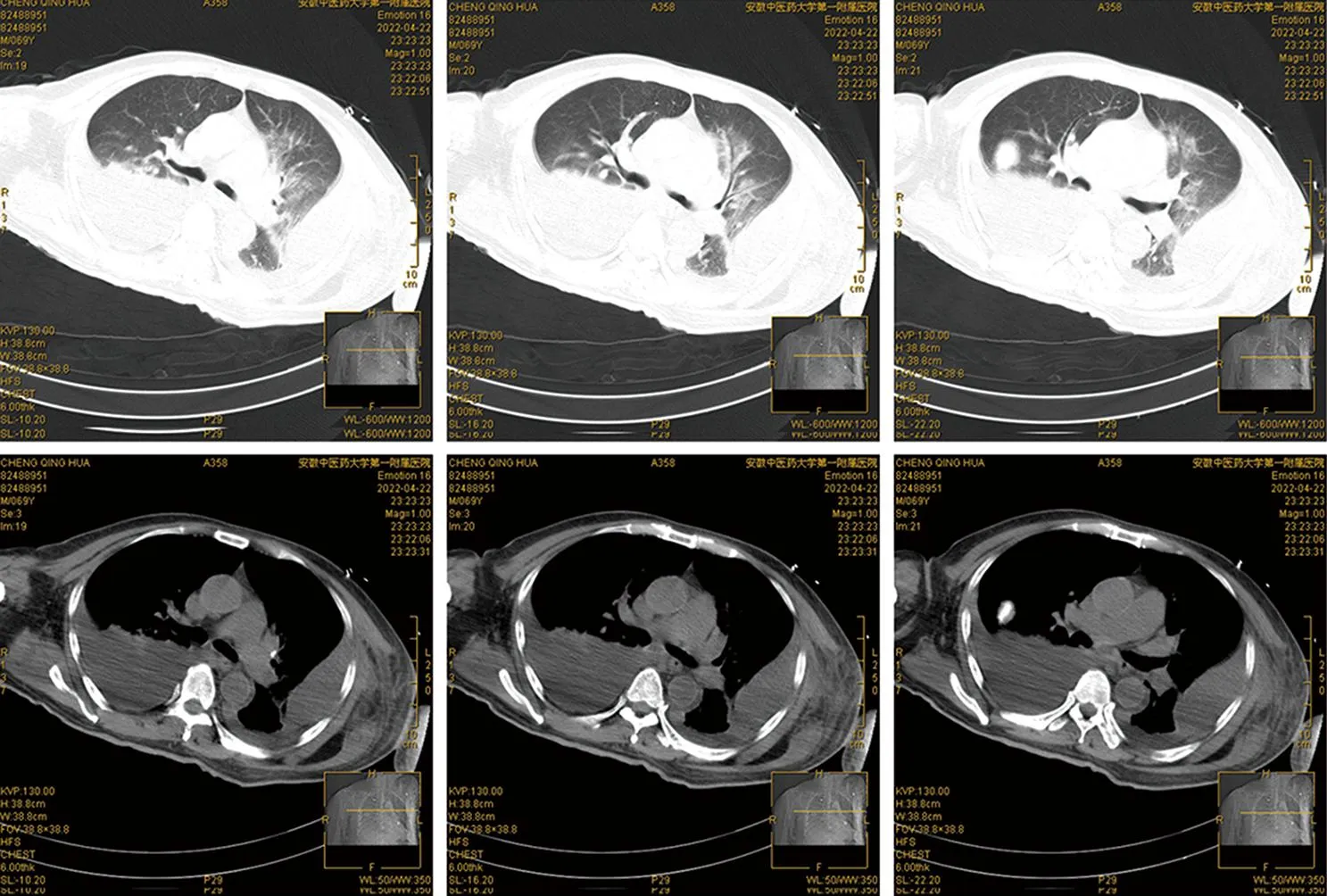
圖2 入院胸部CT影像
1.3 4月23日—25日初始治療經過入科后予抗細菌感染、機械通氣、擴容補液、維持血壓、營養支持等對癥治療。同時予以完善風濕系列、抗中性粒細胞胞漿抗體譜(ANCA)及自身免疫抗體等檢查,均未見異常。行纖維支氣管鏡檢查,灌洗液mNGS示(見圖3):白念珠菌(序列數35)、肺炎克雷伯菌(序列數14)。結合藥敏試驗予以加用伏立康唑抗真菌治療,經治療后,患者體溫高峰逐步降低,炎癥指標逐漸下降,循環趨于穩定,血管活性藥物去甲腎上腺素逐步減撤,但患者血紅蛋白及白細胞仍持續下降,活化部分凝血酶時間(APTT)逐漸延長。
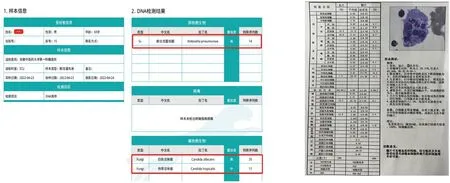
圖3 肺泡灌洗液mNGS檢查結果 圖4 骨髓穿刺檢查結果
1.4 4月26日—5月6日再次診治過程結合患者反復發熱病史、反復查閱患者相關檢查,發現患者鐵蛋白7146.37 ng/mL↑,甘油三酯3.3 mmol/L↑,不排除噬血細胞綜合征可能。予以外送檢查NK細胞活性,結果顯示為1.8%↓,sCD25可溶性白介素2受體8570 U/mL↑,高度可疑噬血細胞綜合征。遂予以完善骨髓穿刺,骨髓細胞檢查提示(見圖4):骨髓增生活躍;粒系增生,粒系占38.35%,紅系占48.06%,粒∶紅=0.8∶1;髓系增生,以中性中幼粒及以下階段細胞為主,各階段細胞形態未見異常;紅系增生,以中晚幼紅細胞為主;淋巴及單核細胞形態大致正常;全片可見噬血組織細胞,有吞噬成熟紅細胞、幼紅細胞、白細胞和血小板現象。結合臨床,補充診斷:噬血細胞綜合征。治療上加用人免疫球蛋白聯合甲潑尼龍沖擊治療,并予輸注紅懸液、血漿等對癥治療。患者一般情況逐漸好轉,體溫高峰降至正常范圍,血三系、凝血功能逐漸好轉,炎癥指標進一步下降,全身水腫減輕。并于4月28日脫機拔管,5月6日復查肺部CT炎癥明顯改善(見圖5),患者癥狀基本好轉,予辦理出院。
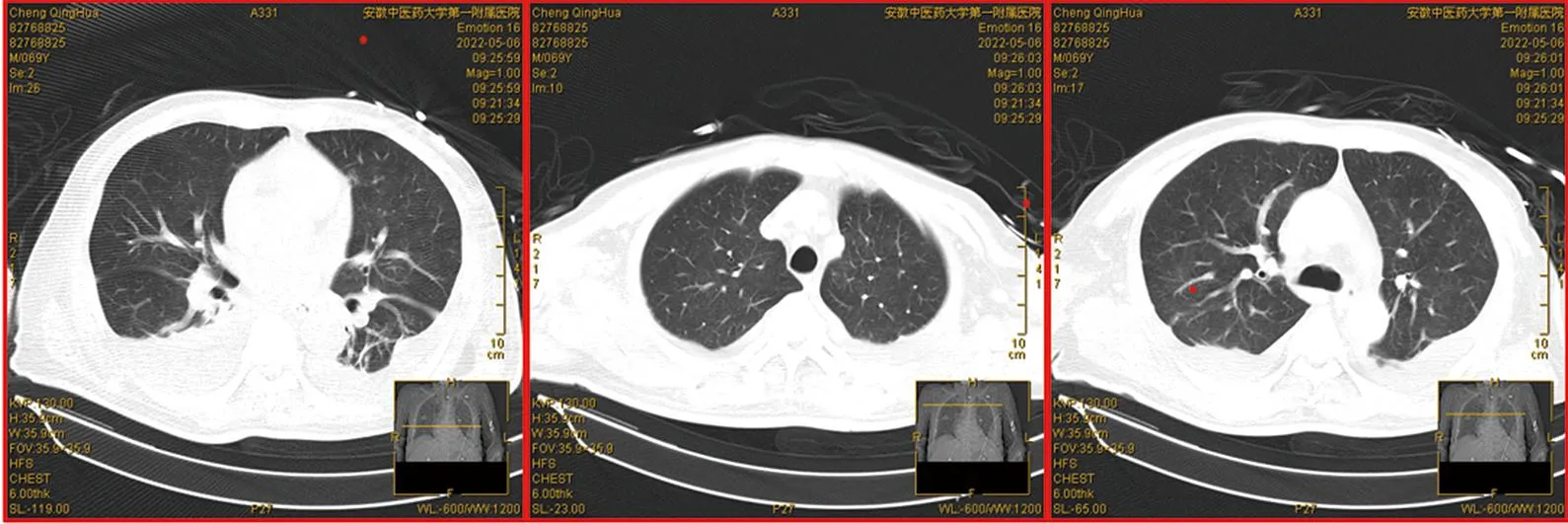
圖5 出院前胸部CT影像
2 討 論
HPS并不是一種單一的疾患,而是一種危及生命的免疫異常激活綜合征,是一種罕見病,主要特點是長期發熱、脾大及三系血細胞減少[3]。一項調查顯示我國HPS總發病率為1.04/10萬,其中兒童(<18歲)為HPS主要發病群體,約占65.40%,成年人則占34.60%[4]。HPS目前認為一般分為原發性(遺傳性)及繼發性(獲得性)兩大類,原發性HPS一般與基因缺陷及免疫缺陷綜合征相關,前者可由一種常染色體或性染色體遺傳至下一代,明確的致病基因為PRF1基因、Munc13-4基因及STX11等[5]。繼發性HPS多由感染、自身性免疫疾病或惡性腫瘤等引起,其中感染相關性HPS與細菌、病毒、真菌及寄生蟲等有關,又以EB病毒引起的最為常見,且其病死率亦是最高[6]。本例患者EB病毒核抗原IgG抗體(EBV-NAIgG)及EB病毒衣殼抗原IgG抗體(EBV-CAIgG)均陽性,考慮患者既往曾經感染過EB病毒,但已好轉,結合檢查考慮與本次HPS相關性不大,治療上未針對性選擇抗EB病毒類藥物及其他抗病毒藥物。
HPS診斷難度極大,曾一度被誤認為是一種幼兒疾病,隨著檢測水平提升,現成人病例報道逐年增多[7]。目前國內仍以國際組織細胞協會HPS-2004方案為診斷依據(符合以下標準中的1條則診斷成立): (1)分子生物學符合HPS。(2)符合以下8條中的5條:①發熱;②脾大;③全血細胞減少,累及≥2個細胞系,血紅蛋白<90 g/L,嬰兒(<4周)血紅蛋白<100 g/L,血小板<100×109/L,中性粒細胞<1.0×109/L;④高甘油三酯血癥和(或)低纖維蛋白原血癥,甘油三酯≥3.0 mmol/L(或≥265 mg/L),纖維蛋白原≤1.5 g/L;⑤骨髓、脾或淋巴結活檢吞噬血細胞現象,無惡性疾病證據;⑥NK細胞活性減低或缺乏;⑦鐵蛋白≥500 mg/mL;⑧可溶性CD25 (或者可溶性IL-2受體)≥2400 U/mL[8]。由于本院檢驗水平限制,未做分子生物學方面檢查,但本病例仍然符合第2條標準中的7條,故可診斷為HPS。
目前針對HPS的治療,大體可分為誘導治療、CNS-HPS治療、挽救治療、維持治療、異基因造血干細胞移植及支持治療等方式,針對感染相關性HPS以抗感染為主[4]。該患者以“發熱待查”入住我科,情況復雜,病因不明,前期考慮重癥肺炎及膿毒癥相關感染性發熱。積極予對癥治療,患者體溫峰值雖有下降,但并未能降至正常水平,全血細胞及凝血功能仍持續惡化,隨時有死亡風險。在及時調整診斷及治療思路,完善相關檢查后,考慮為HPS,與細菌、真菌感染相關可能性大,及時予糖皮質激素聯合人免疫球蛋白沖擊治療,并聯合抗真菌藥物解除病因及加強支持治療,患者病情逐漸好轉出院,隨訪半年病情未再復發。
感染相關性HPS早期并無典型癥狀,其需與普通感染、腫瘤及自身免疫疾病等相鑒別[9]。當發現患者發熱、血二系或三系下降、脾腫大、鐵蛋白異常及合并感染時,應高度警惕感染相關性HPS。本病早期除對癥治療外,應以抗感染控制炎癥風暴為主,還需加用免疫調節相關性藥物,減緩HPS進展,阻止巨噬細胞的再度活化,同時還應關注患者基礎疾病及并發癥,方可改善預后。本文通過我院1例HPS報道并文獻復習,希望可為各同仁提供診斷及治療思路,提高本病診斷率及治愈率。
