
用于高靈敏快速核酸檢測的熒光碳點
2024-01-22 12:11:38常建橋許慧敏謝文菁張洋祁玲范樓珍李勇
物理化學學報 2023年12期
關鍵詞:檢測
常建橋,許慧敏,謝文菁,張洋,祁玲,范樓珍,*,李勇
1北京師范大學化學學院,理論計算光化學與放射性藥物教育部重點實驗室,北京 100875
2中國醫學科學院北京協和醫學院內科,國家癌癥中心/國家癌癥臨床研究中心/癌癥醫院,北京 100021
3中國醫學科學院北京協和醫學院胸外科,國家癌癥中心/國家癌癥臨床研究中心/癌癥醫院,北京 100021
1 引言
PCR技術是指通過多種酶的作用,雙鏈DNA在高溫下變性為單鏈DNA。根據互補堿基配對原則,利用DNA聚合酶從單鏈DNA復制出新的DNA,實現DNA體外擴增1,2。逆轉錄PCR (RTPCR)是指當體系內核酸為RNA時,通過逆轉錄酶將RNA逆轉錄成cDNA,再進行體外擴增的過程。實時定量PCR (qPCR)是在PCR的基礎上,向體系中加入染料或熒光探針,使擴增后的DNA具有熒光性,結合熒光信號強度與擴增增量的關系,可以在擴增過程中實時監測目的DNA的拷貝數3–6。自從新冠病毒流行以來,所采用的標準核酸檢測技術為RT-qPCR技術,其將RT-PCR技術與qPCR相結合,盡管具有強大的檢測性能,但其作為新冠病毒的診斷技術仍然存在一些局限性,例如需要相對較長的檢測過程(2–4 h),專業的設備(熱循環儀、PCR儀等)、實驗室以及專業的操作人員等7–9。因此,開發出新冠病毒快速、靈敏的即時檢測(POCT)是非常必要的。
快速抗原檢測是一種針對特定抗原的POCT,其主要是基于橫向流動免疫分析(LFIA),當含有抗原的樣品滴入到樣品墊后,流到結合物釋放墊與抗體1結合,抗體1上攜帶有色顆粒(例如膠體金),抗原-抗體1-金復合物流向試紙條的檢測區。檢測區是一種多孔膜,存在測試線和控制線。當復合物經過測試線時,抗原與測試線上的抗體2結合,復合物留在測試線,金顆粒發生響應,顯示陽性。而控制線上的響應則表明正確的液體流過試紙條。抗原檢測可提供早期病毒感染的證據10,11,具有成本低、操作方法簡單、檢測時間短(20 min或更短)等優勢12。但是其檢測結果可能會受到其他具有相似結構的化學物質的影響,導致其靈敏度為75%–98%,特異性為95%–99%,容易出現假陽性結果13。由于病毒載量的影響,LFIA的檢測限為1.12 × 105–3.57 × 106copys?mL-114,只能在出現癥狀的第一周有效,容易導致假陰性結果15。抗原檢測這種相對較低的靈敏度給檢測帶來了多重障礙,需要進一步研究以提高其靈敏度和準確性16–18。
碳點(CDs)是一種零維碳納米材料,具有優異的光學性質、良好的水溶性、較好的生物相容性以及廣泛的原料來源等諸多優點19–23,已經應用于檢測24–26、診斷27–29以及藥物的遞送等領域30–32。本研究使用鄰苯二胺和精氨酸作為前驅體,合成了一種基于胍基修飾的熒光碳點(GCDs)(如圖1a)。當GCDs與分子信標(Beacon)通過氫鍵結合后,GCDs的熒光被Beacon所修飾的熒光基團淬滅。遇到目標核酸(Target DNA)之后,Target DNA通過堿基互補配對,與GCDs競爭Beacon分子,GCDs與Beacon脫離,其自身熒光恢復,根據熒光恢復的強度來判斷體系內Target DNA的存在(圖1b),基于這種靈敏的熒光“off-on”,可實現在5 min內快速準確的核酸檢測,并可檢測到體系內0.005 fmol?L-1(約300 copys?mL-1)的核酸序列,我們預計,新建立的COVID-19低成本無擴增檢測將有助于開發新的核酸檢測平臺,以實現COVID-19和其他病原體的高靈敏度和高特異性的快速即時檢驗。

圖1 (a) GCDs合成過程,(b) GCDs核酸檢測機理Fig.1 (a) GCDs synthesis process and (b) nucleic acid detection mechanism of GCDs.
2 實驗部分
2.1 實驗材料和實驗儀器
鄰苯二胺和精氨酸購自上海阿拉丁生化科技股份有限公司,純度為99%。二氯甲烷和甲醇純度為99%,柱層析硅膠和中性三氧化二鋁購自山西諾泰生物科技有限公司。磷酸鹽緩沖液(PBS)、0.22 μm濾膜和7000 kD透析袋購自北京科龍生物科技有限公司。實驗所使用的核酸序列購自上海生工生物有限公司,主要核酸序列見表1。

表1 主要核酸序列Table 1 Major nucleic acid sequence.
熒光光譜儀(法國Horiba Jobin Yvon公司,Fluorolog-3)、透射電子顯微鏡(美國賽默飛世爾科技公司,Talos F200S)、傅里葉變換紅外光譜儀(日本島津公司,IRAffinity-1)、X射線光電子能譜(美國Thermo Fisher公司,ESCALAB 250Xi)、拉曼光譜儀(法國Horiba Jobin Yvon,LabRAM)。
2.2 GCDs的合成
向60 mL去離子水中加入0.15 g鄰苯二胺和0.3 g精氨酸,超聲15 min使其完全溶解。將內膽置于高壓反應釜中,180 °C反應5 h,反應結束后自然冷卻。使用孔徑為0.22 μm的水系濾膜對產物進行過濾。將過濾后的樣品烘干,以甲醇:二氯甲烷(9 : 1)混合溶劑作為洗脫劑,使用200–300目的硅膠和中性氧化鋁進行兩次提純,收集黃色熒光碳點。將得到的產物使用旋轉蒸發儀去除有機溶劑后溶解在水中,得到GCDs溶液。
2.3 GCDs胍基數目的測定
應用坂口反應來對GCDs表面的胍基數目進行測定。取50 μL不同濃度的精氨酸溶液,向其中加入10 μL NaOH溶液,再加入10 μL α-萘酚溶液,混合均勻后加入10 μL 10%的次氯酸鈉溶液,靜置5 min后測量體系內500 nm處的吸收值,繪制精氨酸標準曲線,得到吸收光強度與精氨酸胍基數目的對應關系。按照相同的方法對GCDs中的坂口反應吸收值進行測量,對照標準曲線得到GCDs的胍基數目。
2.4 GCDs-Bea的制備
將GCDs與Beacon在PBS中以不同比例混勻后,渦旋震動10 s,室溫避光靜置15 min,然后使用7000 kD的透析袋進行透析2–3天,除去未與Beacon結合的游離GCDs,取出透析袋內液得到GCDs-Beacon (GCDs-Bea)。
使用JY-SPBT電泳設備對GCDs-Bea進行瓊脂糖凝膠電泳分析。凝膠濃度2%,電壓85 V,電泳時間40 min,Running Buffer為TAE緩沖液。電泳結束后,使用凝膠成像儀在254 nm激發波長下對瓊脂糖凝膠進行成像,使用Image J軟件對凝膠成像結果進行灰度值分析。
2.5 核酸檢測
向2 mL GCDs-Bea中分別加入100 μL的PBS,Target DNA,Mismatch DNA等核酸序列,在室溫混勻后使用熒光光譜儀測量體系內熒光光譜,并對570 nm處的熒光變化率(F-F0)/F0×100%進行分析。
3 結果與討論
3.1 GCDs結構表征
GCDs的結構表征如圖2所示,圖2a為GCDs的TEM圖像,GCDs具有均勻的分散狀態,其平均粒徑約為4 nm,大小均一,高分辨TEM圖顯示GCDs的晶格間距為0.21 nm,對應于石墨的(100)晶面的衍射。圖2b為傅里葉變換紅外光譜(FT-IR)結果表征,在3433 cm-1處有對應N―H的振動峰,表明GCDs表面存在―NH2基團。在3172 cm-1處有對應羧酸―OH的振動峰,表明GCDs表面存在―COOH基團,1506和1637 cm-1處有兩個強峰,這是C=C/C=N的伸縮振動特征峰。通過坂口反應測定后,對應精氨酸濃度與吸收值的標準曲線,如圖2c所示,計算表明一個GCDs上約帶有3–4個胍基。為了進一步探究GCDs的結構,分別進行了X射線光電子能譜(XPS)和拉曼光譜的表征。如圖2d所示,用XPS確定了GCDs的元素組成。將XPS數據擬合后得到高分辨譜圖,如圖2e的高分辨C 1s光譜顯示,GCDs中碳元素主要由C=C/C―C (284.6 eV)、C―N/C―O (285.1 eV)和C=O (288.3 eV)形式構成。圖2f拉曼光譜進一步表征了GCDs的結構,在1385 cm-1(D峰,碳材料中的無序態)和1597 cm-1(G峰,碳材料中的結晶態)附近有兩個寬峰,分別對應碳原子的sp3和sp2雜化振動。GCDs的ID/IG約0.6,這表明所制備的GCDs具有高度的晶體結構。這一結果也與HRTEM結果一致。綜上所述,GCDs是中心為較好共軛性的剛性碳核結構,邊緣具有―NH2、―COOH以及3–4個―C3H4基團的熒光碳點(圖1a)。

圖2 GCDs的(a) TEM圖,高分辨TEM圖以及尺寸統計分布圖,(b) FT-IR圖,(c)坂口反應標準曲線,(d) XPS全譜,(e) C 1s高分辨譜圖,(f)拉曼圖Fig.2 (a) TEM,high resolution TEM and size statistical distribution diagram,(b) FT-IR,(c) standard Sakakuchi reaction curve,(d) XPS full spectrum,(e) C 1s high resolution spectrum,(f) Raman diagram of GCDs.
3.2 GCDs的光學性質
圖3a是GCDs的紫外可見吸收光譜圖,GCDs在波長為210 nm處存在明顯的吸收峰,對應于sp2共軛結構的π–π*的躍遷吸收。在波長為258 nm附近存在幾個較弱吸收峰,對應于C=O等結構的n–π*躍遷吸收。GCDs在415 nm處有特征吸收峰,對應的熒光發射峰位置為570 nm。圖3a插圖為GCDs在日光下的照片和紫外光照射下的熒光圖像。使用積分球測得GCDs在410 nm 激發下的熒光量子產率為9.8%。圖3b是GCDs的熒光激發光譜和發射光譜,GCDs的發射光譜從激發光波長400 nm增加到520 nm時,最大發射峰均在570 nm,不存在激發依賴現象,與圖3c的二維熒光光譜圖對應。并且GCDs室溫存放17個月后,熒光發射位置不發生移動,熒光強度下降約4.5%,如圖3d所示,GCDs的發光穩定性較好。
3.3 GCDs-Bea的制備
圖4a為瓊脂糖凝膠電泳阻滯實驗結果,Beacon與凝膠中的GelRed結合后,在254 nm激發下產生熒光條帶,經過凝膠成像系統拍照后,使用Image J軟件測量條帶灰度值,結果表明,向Beacon中加入GCDs后,Beacon亮度減弱,GCDs與Beacon發生相互作用,阻礙了Beacon條帶的移動,當向Beacon中加入2倍量GCDs時,Beacon條帶幾乎消失(圖4a左一),過量的GCDs與Beacon發生相互作用后完全阻滯了Beacon的移動。

圖4 GCDs與Beacon連接結果。(a)瓊脂糖凝膠電泳條帶的灰度值,(b) GCDs上胍基基團與Beacon四種堿基相互作用能量Fig.4 GCDs-Bea connection result.(a) Gray value of bands in agarose gel electrophoresis,(b) calculation results of guanidine groups and beacon bases.
對GCDs的結構進行簡化后計算GCDs與Beacon的結合能。使用幾何建模方法確定了GCDs中的氨基(―NH2)、羧基(―COOH)和胍基(―CN3H4)與腺嘌呤(A)、鳥嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T)四種堿基之間的相互作用,并在b3lyp/6-31g(d)水平上優化了幾何結構,通過從頭算量子化學方法計算了這些模型的相互作用能量33。如圖4b的計算結果表明,胍基與Beacon的相互作用能量為-1.25 – -7.01 kcal?mol-1(1 kcal?mol-1=4.1868 kJ?mol-1),而氨基和羧基與Beacon的相互作用能量相似(分別為0.58 – -2.21 kcal?mol-1和2.55 – -3.07 kcal?mol-1),表明胍基在與Beacon相互作用時具有明顯的能量優勢。
3.4 核酸檢測結果的特異性與靈敏度
向GCDs-Bea (Control)中分別加入Target DNA以及錯配DNA (Mismatch DNA,序列與Target DNA不同),結果如圖5a所示。GCDs-Bea與Target DNA混勻后測量熒光光譜,熒光強度增強6倍,但Mismatch DNA組的熒光沒有發生明顯變化,顯示GCDs-Bea核酸檢測具有較強的特異性。為了明確檢測方法的特異性,設置了55個樣本,其中34個為含有不同濃度的Target DNA的陽性樣本,21個含有不同序列的Mismatch DNA的陰性樣本,使用GCDs-Bea對55個樣本進行熒光測定并分析,檢測閾值設置為熒光強度變化率為20%。結果如圖5b,陽性樣本平均熒光增長率為124.75%,陰性樣本平均熒光增長率為0.04%。其中,陰性組存在一樣本熒光增長率為20.35%,為假陽性結果。所以GCDs-Bea的核酸檢測特異性=真陰性樣本/(真陰性樣本+假陽性樣本) × 100% = 95.45%。

圖5 GCDs-Bea核酸檢測的(a)特異性,(b)多樣本特異性,(c)靈敏度。圖c中的濃度為檢測時的起始濃度,終濃度為該濃度的5%Fig.5 (a) Specificity,(b) multiple sample,(c) sensitivity of GCDs-Bea nucleic acid detection. The concentration in the figure c is the initial concentration at the time of detection,and the final concentration is 5% of that concentration.
為了確定檢測的靈敏度,將GCDs-Bea與不同濃度的Target DNA進行孵育,測量分析GCDs的熒光變化,圖5c顯示出GCDs-Bea對不同濃度的Target DNA產生不同的熒光現象,DNA濃度即使低至0.01 fmol?L-1(終濃度約為300 copys?mL-1)時,仍然有熒光增加現象,表明了GCDs-Bea具有較強的靈敏度。對比RT-qPCR檢測方法的檢測限2.59 × 102–1.04 × 103copys?mL-1,快速抗原檢測試劑盒的1.12 × 105–3.57 × 106copys?mL-114,GCDs-Bea的檢測方法可以靈敏地檢測出目標核酸序列,而不需要進行核酸擴增步驟。此外,GCDs-Bea可以在兩周內保持穩定,不影響檢測效果,將DNA加入到GCDs-Bea后,可立即進行熒光測定,獲得檢測結果,該過程預計在5 min內。
但是在檢測過程中發現,熒光恢復的強度與Target DNA的濃度不呈線性關系,推測原因與GCDs-Bea的連接機理有關。GCDs與Beacon連接的數量比并不一致,所以Target DNA與Beacon特異性結合后,脫離下來的GCDs數量不一致,恢復的GCDs的熒光有所不同,導致DNA濃度增大后,熒光值可能減小。但是可以確定的是,熒光值雖然減小,但是依然比Control組高20%以上。
3.5 其他核酸序列的檢測
為了明確GCDs-Bea體系能否檢測其他病毒或者疾病,將GCDs與不同序列的Beacon分子連接(Beacon2和Beacon3),然后將其與對應序列Target DNA2和Target DNA3進行測試,如圖6a–c所示,更換Beacon核酸序列后,GCDs-Bea在檢測到Target DNA后,GCDs的熒光強度變化率均在50%以上。將Target DNA的核酸序列改變一個堿基(Mis1)或4個堿基(Mis4),再分別與GCDs-Bea2反應,如圖6d所示,當GCDs-Bea體系內存在Target DNA,熒光強度變化率為24%,改變Target DNA中的一個堿基,熒光強度變化率降為13%,改變4個堿基后,變化率僅為3%,與Mismatch DNA相同。檢測體系具有與Beacon1相似的特異性和靈敏度。實驗表明GCDs-Bea檢測體系可以通過改變Beacon的核酸序列,進行不同病毒或疾病的核酸檢測。
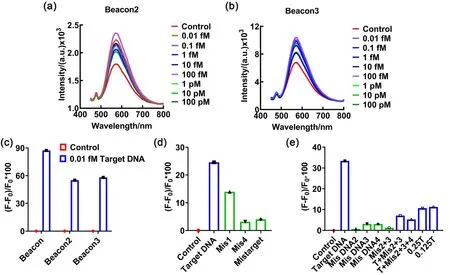
圖6 GCDs和不同核酸序列Beacon2 (a)和Beacon3 (b)的靈敏度結果,(c) Beacon、Beacon2和Beacon3的熒光檢測結果,(d) GCDs-Bea2的核酸檢測的特異性,(e)混合樣品的核酸檢測。圖中的濃度為檢測時的起始濃度,終濃度為該濃度的5%Fig.6 The sensitivity results of GCDs and different nucleic acid sequences of Beacon2 (a) and Beacon3 (b),(c) fluorescence results of Beacon,Beacon2 and Beacon3,(d) specificity of nucleic acid detection of GCDs-Bea2,(e) the nucleic acid detection of mixed samples.The concentration in the figure is the initial concentration at the time of detection,and the final concentration is 5% of that concentration.
保持核酸終濃度一致,將不同序列的核酸進行混合后,GCDs-Bea依然能檢測到其中含有的微量Target DNA,如圖6e所示,MisDNA2、3、4分別為改變Target DNA中的2、3、4個堿基,T +Mis2 + 3 + 4為Target DNA、MisDNA2、MisDNA3和MisDNA4混合物。0.25T為0.25倍的0.01 fmol?L-1Target DNA,即向體系內加入0.0025 fmol?L-1Target DNA。所以當體系內存在多種核酸甚至整個病毒碎片時,依然可以對其中含有的微量目標核酸進行檢測,即對獲得的咽拭子或鼻拭子采集液進行快速的病毒破壞后,GCDs-Bea與采集液進行混合后,依然能對目標核酸進行靈敏地檢測,避免了核酸提純這一工序,節省了檢測時間。例如現有的核酸檢測需要在采集樣本后,在實驗室進行30 min的紫外滅菌,再進行一系列地核酸提純、擴增、檢測(2–4 h),所以GCDs-Bea核酸檢測可能為之后的檢測提供新的便利。
4 結論
本研究成功合成了一種基于胍基修飾的熒光碳點(GCDs),連接分子信標(Beacon)后,可以在5 min內識別出目標核酸序列,并可檢測到體系內0.005 fmol?L-1(約300 copys?mL-1)的核酸序列,不需要進行核酸擴增過程,并且可以在混合體系內識別出目標核酸序列,不需要進行核酸提取過程。具體過程主要為,當GCDs與Beacon結合后,GCDs與Beacon所攜帶的ROX熒光基團發生熒光共振能量轉移,GCDs熒光被淬滅,GCDs-Bea熒光減弱,但當GCDs-Bea遇到與Beacon互補配對的目標核酸序列(Target DNA)時,Beacon與GCDs分離,GCDs熒光恢復。實驗結果表明相比于目前的RT-qPCR檢測方案,該方法可以縮短檢測時間,提高檢測效率。GCDs-Bea可以通過改變Beacon的核酸序列的方式,來達到對其他病毒或疾病的核酸檢測的結果。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48

- 物理化學學報的其它文章
- Performance Improvement and Antibacterial Mechanism of BiOI/ZnO Nanocomposites as Antibacterial Agent under Visible Light
- 2D/3D S-Scheme Heterojunction Interface of CeO2-Cu2O Promotes Ordered Charge Transfer for Efficient Photocatalytic Hydrogen Evolution
- Construction of a Highly Active Rh/CeO2-ZrO2-Al2O3 Catalyst Based on Rh Micro-Chemical State Regulation and Its Three-Way Catalytic Activity
- Pickering Emulsion Templated Proteinaceous Microsphere with Bio-Stimuli Responsiveness
- Electronic Modulation of Ni-Mo-O Porous Nanorods by Co Doping for Selective Oxidation of 5-Hydroxymethylfurfural Coupled with Hydrogen Evolution
- RuP Nanoparticles Anchored on N-doped Graphene Aerogels for Hydrazine Oxidation-Boosted Hydrogen Production