模仿IgG4 相關淋巴結病的多中心型Castleman 病:易誤診病例的鑒別診斷及文獻復習
2024-03-10 13:42:36李春艷劉國榮常麗君李秀博丁文雙
廣州醫藥 2024年2期
李春艷 劉國榮 常麗君 李秀博 丁文雙
1 廣州市第一人民醫院,華南理工大學第二附屬醫院病理科(廣東廣州 510180)
2 南方醫科大學附屬茂名醫院病理科(廣東茂名 525000)
Castleman病(Castleman’ disease,CD)是一種罕見的淋巴增生性疾病,根據臨床表現分為單中心型(unicentric Castleman’ disease,UCD)和多中心型(multicentric Castleman’s disease,MCD)[1]。MCD根據是否感染卡波西肉瘤相關皰疹病毒/人類皰疹病毒8(Kaposi’s sarcoma-associated herpesvirus,KSHV/human herpesvirus-8,HHV8)分為KSHV/HHV8感染相關性、特發性和POEMS相關MCD(POEMS-MCD)。特發性多中心型Castleman病(idiopathic MCD,iMCD)分為漿細胞型(plasma cell type-iMCD,PC-iMCD)及高血管型(hyaline-vascular-iMCD,HV-iMCD),其中以PC-iMCD多見,PC-iMCD的淋巴結表現為淋巴濾泡增生、生發中心萎縮,濾泡間區成熟漿細胞片狀增生[2-3]。IgG4相關淋巴結病有多種生長模式,其組織形態不具有特異性,其中I型生長模式與PC-iMCD有較多重疊之處,PC-iMCD可伴有大量IgG4陽性漿細胞浸潤且血清學IgG4水平異常升高,當PC-iMCD形態及實驗室檢查同時符合IgG4相關淋巴結病診斷標準時,兩者的鑒別非常困難,但兩類疾病的治療方案及預后完全不同,必須將其區分[4-5],這給病理診斷帶來很大挑戰,容易造成誤診。本文旨在分享1例與IgG4相關性淋巴結病的組織病理學相似的PC-iMCD,結合其臨床癥狀、影像學特征及實驗室檢查并回顧相關文獻以探討2種疾病的鑒別要點,旨在加強臨床醫師對兩種疾病的認識,避免誤診、誤治。
1 資料與方法
1.1 病例資料
患者為48歲女性,因口干、多飲18年,皮膚瘙癢2年,加重1個月入院。既往有貧血、2型糖尿病病史。體格檢查:未見明顯異常。實驗室檢查顯示多項體液免疫指標升高:IgA 13.4 g/L;IgG 91 g/L;IgM 2.57 g/L;IgE >6 000 ng/mL;補體C3 1.2 g/L;補體C4 0.239 g/L;Kappa輕鏈2 230.0 mg/dL;Lambda輕鏈1 290.0 mg/dL;血清IgG4水平14.7 g/L。胸部CT平掃及多平面重建:雙側腋窩、縱隔、上腹部腹膜后多發腫大淋巴結,考慮血液系統疾病可能;脾臟多發占位,性質待定。PETCT:全身多發淋巴結代謝稍活躍,全身多處骨髓彌漫性代謝稍活躍,考慮淋巴瘤多發浸潤,建議結合病理檢查;脾大伴有稍高密度結節,代謝不高,疑浸潤。
1.2 蘇木素-伊紅(HE)染色
4%中性甲醛液固定標本,石蠟包埋切片(厚度=3 μm)經脫蠟、水化后用全自動HE染片機(賽默飛,型號:Shandon Varistain Gemini)行HE染色。
1.3 免疫組織化學(免疫組化)染色及EB病毒原位雜交
采用4%中性甲醛液固定標本,石蠟包埋切片(厚度=3 μm)常規脫蠟水化后,采用Leica全自動免疫組化染色機(設備型號:BenchMark ULTRA型)進行免疫組織化學染色。本研究標記抗體包括CD3、CD5、CD20、CD79a、CD10、CD21、CD56、CD38、CD138、CD43、Bcl-2(B cell lymphoma protein-2)、Bcl-6、MUM-1[Multiple myeloma(MM)antigen 1)]、S-100、IgG4、IgG、Ki-67,上述抗體均為即用型抗體,購自廣州安必平醫藥科技股份有限公司,均設置陽性對照。EB病毒原位雜交試劑盒購于北京中杉金橋生物技術有限公司,根據試劑盒步驟進操作,同時設置陽性對照。
2 結 果
2.1 鏡下HE形態
淋巴濾泡增生,生發中心萎縮(圖1A),部分套區淋巴細胞呈“洋蔥皮”樣圍繞生發中心排列(圖1B),濾泡間區擴張伴血管增生,其內見大量成熟的漿細胞呈片狀浸潤,灶區見含鐵血黃素沉積(圖1C-D),漿細胞及淋巴細胞均未見明顯異型性。
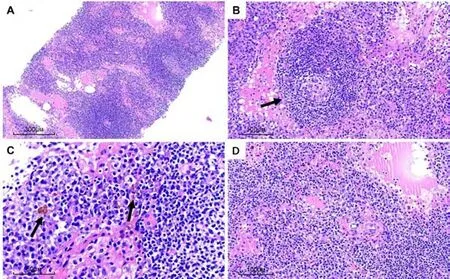
圖1 PC-iMCD 鏡下組織形態
2.2 免疫表型
濾泡樹突網CD21陽性,顯示生發中心萎縮(圖2A);濾泡間區成熟漿細胞CD79α、CD38、CD138(圖2B)、MUM1及CD43陽性;濾泡間區IgG4陽性漿細胞>100/高倍視野,IgG4陽性細胞數/ IgG陽性細胞數比值>40%(圖2C-D);Kappa及Lambda輕鏈顯示非限制性表達(圖2E、F);生發中心B淋巴細胞CD20、CD79α、CD10及Bcl-6陽性,Ki-67增殖指數20%;T淋巴細胞CD3、CD5陽性。
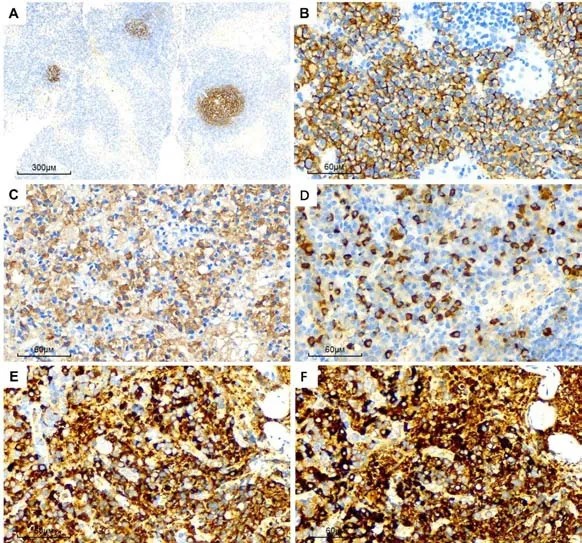
圖2 PC-iMCD 免疫表型
2.3 EB病毒檢測
EB病毒編碼RNA原位雜交陰性(圖3A)。

圖3 PC-iMCD 原位雜交
3 討 論
本研究報道了1例少見PC-iMCD的淋巴結病變,其組織學特征與IgG4相關淋巴結病變-MCD樣亞型組織學改變類似,鏡下均表現為淋巴濾泡增生,濾泡間見大量成熟漿細胞浸潤。同時該患者淋巴結內IgG4計數>100/高倍視野、IgG4/IgG比例>40%、血清IgG4水平14.7 g/L(IgG4相關疾病臨界值1.35 g/L),其形態學、免疫組化及實驗室檢查均符合IgG4相關淋巴結病變診斷標準。兩種疾病的治療措施及生存預后有較大差異,在此情況下,兩類疾病的的鑒別極為重要,結合文獻復習所總結的鑒別要點見表1。
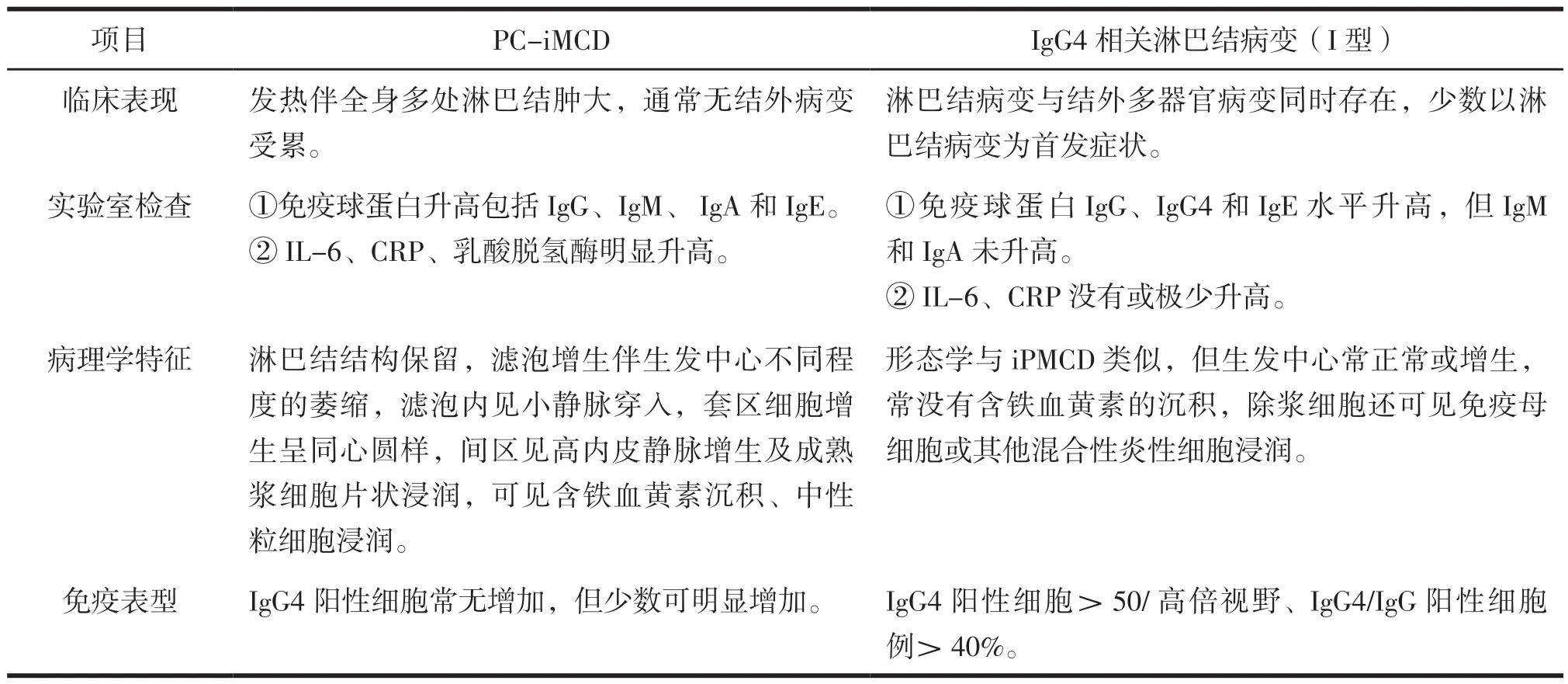
表1 PC-iMCD與IgG4相關淋巴結病變(I型)的鑒別總結
IgG4相關病變(IgG4-related disease,IgG4-RD)為系統性疾病,臨床表現全身多處器官受累,最常累及胰腺、唾液腺、淚腺、腮腺和肺[3]。IgG4-RD確診依靠綜合性診斷,修訂版(2020版)診斷標準[6]中包括臨床影像、血清學和病理診斷標準,其中特征性的病理改變包括:①受累組織內大量淋巴漿細胞浸潤,伴纖維化;②IgG4+漿細胞/IgG+漿細胞比值>40%,且IgG4+漿細胞>10/高倍視野(診斷淋巴結病變需>50/高倍視野[7]);③席紋狀纖維化和閉塞性靜脈炎,其中IgG4+漿細胞絕對值需與器官特異性IgG4-RD 診斷標準中臨界值相結合。80%的IgG4-RD伴全身多處淋巴結腫大,大部分IgG4相關淋巴結病變與結外病變同時存在,僅少數病例以淋巴結病變為首發癥狀,隨后出現典型結外組織學改變[8]。目前IgG4相關的淋巴結病變分為5種組織學類型,多中心Castleman病樣(Ⅰ型)、濾泡增生(Ⅱ型)、濾泡間擴張(Ⅲ型)、生發中心進行性轉化(Ⅳ型)和炎性假瘤樣(Ⅴ型)[4,9]。IgG4相關淋巴結病變組織學不具特異性,如I型淋巴結改變與MCD相似,鏡下均表現為淋巴結結構保留,髓竇擴張開放,濾泡增生伴生發中心不同程度的萎縮退變,濾泡內見小靜脈穿入,濾泡間區可見高內皮靜脈增生及成熟漿細胞大量浸潤[4]。值得注意的是,IgG4+漿細胞浸潤并非IgG4相關淋巴結病變特有,部分自身免疫性疾病相關淋巴結病變,MCD等也可出現IgG4+漿細胞大量增生,少數情況下可伴血清中IgG4水平升高,因此診斷IgG4相關淋巴結病變需與伴有類似組織學和血清學改變的疾病進行鑒別[6]。
C D 是一種罕見的淋巴增殖性疾病,依據臨床表現分為單中心型(U C D)和多中心型(MCD)。不伴KSHV/HHV8感染的MCD稱之為iMCD,iMCD包括PC-iMCD和HV-iMCD。PCiMCD發病與IL-6異常升高有關,患者表現為發熱伴全身多處淋巴結腫大、血清蛋白異常升高、高乳酸脫氫酶等,鏡下改變主要為淋巴濾泡增生伴生發中心萎縮,部分生發中心可見小靜脈插入,髓竇擴張,濾泡間區大量成熟漿細胞片狀浸潤[10]。部分PC-iMCD患者淋巴結病變可伴有IgG4+漿細胞浸潤及血清IgG4水平升高,韓國一項納入87例CD的研究發現,11.5%(10/87)病例可滿足IgG4相關淋巴結病變診斷標準[11]。Sato等[12-13]分析總結IgG4相關淋巴結病變與MCD組織學鑒別要點如下:①MCD患者生發中心常萎縮,IgG4相關淋巴結病變則正常或增生;②MCD濾泡間區成熟漿細胞為片狀增生模式,IgG4相關淋巴結病變則細胞成分混雜,表現為成熟漿細胞與漿樣細胞及免疫母細胞的增生;③IgG4相關淋巴結病變內可見嗜酸性粒細胞浸潤,MCD未見。
為了更加精準的診斷IgG4相關病變,日本學者Sato等[14]提出了IgG4-RD的排除性診斷標準,目的在于排除PC-iMCD等具有相似組織學改變的疾病。該排除性診斷標準臨床特征包括:①血清CRP持續升高(≥10 g/L);②血清IgA升高;③血清IgM升高。病理特征包括:①漿細胞片狀增生;②鐵血黃素沉積;③中性粒細胞浸潤。隨后,Nishikori等[3]通過納入58例PC-iMCD的資料進一步驗證此排除標準,發現其中20.5%(8/39)淋巴結病變和42.1%(8/19)肺病變可符合IgG4-RD診斷標準,通過排除性診斷標準,16例患者均可排除IgG4-RD,該排除性診斷標準的靈敏度和特異度分別為100%和93.1%。結合該排除性診斷標準,本文報道的病例可排除IgG4相關淋巴結病變,符合PC-iMCD的診斷標準。
Xia等[15]把組織學改變同時滿足IgG4-RD和MCD診斷標準的這類少見病變創新給予IgG4-CD暫定名稱,作者從臨床表現、實驗室檢查及臨床治療預后角度進行了回顧性分析。IgG4-CD病例少見,其發病率約占IgG4-RD的2.5%(15/534),IgG4-CD發病部位兼具MCD和IgG4-RD的特征。本組數據中全部IgG4-CD病例(15/15)累及多處淋巴結,同時87%病例(13/15)伴淋巴結外多部位累及,常見于舌下腺、腮腺、胰腺及副鼻竇等部位。與MCD相比,IgG4-CD血紅蛋白、嗜酸性粒細胞數量、血清IgG4水平及IgG4/IgG比例偏高,而CRP、IL-6水平等偏低。IgG4-CD患者可接受單純糖皮質激素治療或聯合糖皮質激素/免疫抑制劑治療,該類患者治療效果及預后同IgG4-RD較一致,生存預后明顯優于MCD。該作者首次創新性地從臨床及治療角度提出IgG4-CD疾病名稱,為解決這類難以鑒別的疾病提供了新的診斷和治療思路,但該組數據的病理學資料描述不夠詳盡,同時受樣本量少的限制,該暫定名稱的疾病在病理學及風濕免疫學領域的接受度可能受到挑戰。
伴血清IgG4升高的PC-iMCD與IgG4相關淋巴結病(Ⅰ型)組織形態、免疫表型特征及血清學改變均有重疊,單純依靠形態學難以將兩者進行鑒別,容易造成誤診、誤治。兩者鑒別需結合IgG4相關病變診斷標準、排除性診斷標準、臨床表現及相關實驗室檢查綜合判斷。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
