
高效液相色譜-串聯質譜法測定精油類化妝品中生物堿研究
2024-03-20 07:33:26潘廣樂
山西化工 2024年2期
潘廣樂
(廣州市勝蔻生物科技有限公司,廣東 廣州 510990)
0 引言
精油類化妝品在經過蒸餾、溶劑提取后,可濃縮獲取植物中多種人體有利成分和不利成分。植物精油的沸點范圍一般為150~300 ℃,其實際提取過程中會殘留一部分生物堿,其中有些生物堿具有一定毒性,如鉤吻堿、烏頭堿等,因而必須提高精油類化妝品安全問題的相關重視。現有研究中針對化妝品中生物堿測定的研究成果較多,但相關成果大多集中在水劑、乳液膏類化妝品,對于精油類化妝品的研究則相對較少。因此,采用高效液相色譜-串聯質譜法對精油類化妝品中生物堿進行檢測分析,介紹一種精油類化妝品中生物堿檢測方法,將具有一定研究價值。
1 實驗儀器與試劑
1.1 實驗儀器
液相色譜-串聯四極桿質譜儀、超聲波清洗器、離心機、快速攪勻器、電子天平等。
1.2 實驗試劑
甲醇(色譜純,分析純)、乙腈(色譜純,分析純)、甲酸(LC-MS 級)、的寧、士延胡索乙素、雷公藤(次堿,吉堿)、青藤堿、秋水仙堿、山梗菜堿、阿托品、麻黃堿、鉤吻素子、黃華堿、西伐丁、那可丁、高三尖杉酯堿、東莨菪堿、山莨菪堿、馬錢子堿、倒千里光堿、蘆竹堿、新/次烏頭堿、毛果蕓香堿、苯甲酰/苯甲酰新/苯甲酰次烏頭原堿、麥角新堿、鉤吻素甲、氧化苦參堿、吳茱萸堿、哈爾堿、毒扁豆堿。
2 標準溶液配制
31 種生物堿標準品用電子天平精確稱量,放入容量瓶中,用甲醇溶液將其分別定容,制成質量濃度100 mg/L 的標準樣品,并將其轉入褐色小瓶,在-20 ℃冷藏。
將標樣溶液準確地吸入后,將其放入容量瓶中,用質量分數為20%的甲醇溶液進行稀釋和定容,制成質量濃度為10 mg/L 的混合標準溶液,并將該溶液轉入褐色小瓶,4 ℃低溫環境保存。
精確吸取10 mg/L 的混合標準溶液,用的甲醇-水溶液(體積比3∶1)稀釋,得到質量濃度為2.5、10、20、50、100、250、500 μg/L 的標準液。另外,吸取10 mg/L 混合標準溶液,采用陰性空白基體萃取液進行稀釋和定容,制得質量濃度為2.5、10、20、50、100、250、500 μg/L 的陰性標準溶液。
3 樣品前處理及實驗條件
3.1 樣品前處理
所用的樣本都是市場上常用的植物精油。用電子天平精確步驟1.0 g 試樣,放入25 mL 帶塞比色管中,按3∶1 的體積比將包含2%(體積分數)甲酸溶液的甲醇-水溶液定容至10 mL。先用快速混合機進行1 min 的混合,然后放入超聲波設備中萃取10 min,最后用吸管添加正己烷1 mL,用快速混合機處理5 min,除去樣本中的脂質。將試樣放入離心機,在4 000 r/min 下離心10 min,移除正己烷層,將上清液用0.22 μm 有機濾膜濾出,用LC-MS/MS 對其進行分析。
3.2 實驗條件
色譜條件:采用HPH-C18 色譜柱,試驗中色譜柱溫度控制為35 ℃;色譜流動相A 采用體積分數為0.2%的甲酸水溶液,色譜流動相B 采用乙腈,通過兩種流動相對色譜柱實施梯度洗脫,洗脫過程中流動相流速和進樣量應分別控制為0.35 mL/min、5 μL。
質譜條件:實驗過程中質譜分析方法為正離子掃描、多反應監測等;質譜分析過程中儀器霧化壓力控制為276 MPa;設備干燥氣通入溫度為300 ℃,干燥氣流速控制為8 L/min;設備毛細管電壓控制為4 000 V。
4 結果與討論
4.1 色譜-質譜條件優化
質譜條件優化:考慮到生物堿中含有的N 原子上的孤電子易與質子結合,所以實際質譜正離子掃描時將采用一級質譜掃描模式,完成掃描后選用分子離子為母體,實施二級質譜掃描,根據掃描結果選取2 個高豐度、低干擾的子離子作為實驗中質譜分析的定量和定性離子,以2 個離子為標準對二級質譜進行優化調整,確保2 個離子響應最大化,由此獲取相應的質譜參數。
色譜條件優化:根據現有研究成果(圖1)可知,相較于HPH-C18 色譜柱,Bonus-RP 色譜柱的總離子流色譜圖中存在多個包峰,說明色譜總體分離效果較差。具體分析后,確認HPH-C18 色譜柱對部分生物堿的鍵合能力不足,以至于色譜柱分析中出現保留效果不足等情況。不過從整體角度來看,色譜柱分離后總離子流色譜圖的峰形較好,應用此色譜柱后可有效增強色譜分析的總體分離度,因而綜合分析后選用HPH-C18 色譜柱作為色譜分析色譜柱。

圖1 HPH-C18 色譜柱對31 種生物堿總離子流色譜圖
4.2 前處理條件優化
提取溶劑優化。分別采用甲醇和乙腈作為提取溶劑,對比分析后確認相較于采用乙腈作為提取溶劑時,采用甲醇作為提取溶劑的響應水平更高,因而選用甲醇作為提取溶劑。當提取溶劑中的有機相為100%時,通過HPH-C18 色譜柱進行色譜分析后發現色譜峰存在明顯前延情況,并且得到的色譜峰峰形較差。因此,在對比不同水與甲醇比例條件下樣品回收率以及色譜峰形變化后,確認水與甲醇之間的體積比為1∶3 時,通過HPH-C18 色譜柱獲取的色譜峰峰形最好,因而將提取溶劑為水與甲醇體積比設置為1∶3。此外,在提取溶劑中加入適量的酸有利于增強生物堿的水溶性,所以在對比提取溶劑中加入不同量甲酸后,確認最佳甲酸添加量為2%(體積分數),此時提取溶液對各種生物堿均有著較高的回收率。
凈化條件優化。精油類化妝品中蘊含有較多的油脂類物質,為保障精油類化妝品中生物堿測定精準性,需要在樣品前處理過程中對油脂進行有效去除。常用的油脂去除溶劑為正己烷,近年來部分學者采用EMR-Lipid 固相萃取小柱進行油脂類物質去除,為確認兩種油脂去除方法的有效性,實施兩種方法對油脂去除效果對比,進而獲取到圖2 中的對比分析結果。根據對比結果可知,相較于正己烷溶劑(HEX 除脂),EMR 固相萃取小柱(EMR 除脂)僅在去除含有5 個以上碳鏈的脂肪類物質中表現出較好的成效,而精油類化妝品中不含有相關物質,因而為有效降低測定成本,選用正己烷溶劑作為凈化溶劑。

圖2 兩種油脂去除方法的效果對比
4.3 基質效應的評價
基質效應通常會影響測定結果的準確度、靈敏度和準確度。以混合標準溶液作為測定樣品,選取陰性標準溶液濃度作為橫坐標軸,離子對峰面積為縱坐標軸,根據試驗數據繪制相應的基質效應曲線。目前已有多種方法可以對基質效應進行評估,其中基質效應的研究主要是利用基質標準溶液的斜率與基體標準曲線的斜率之比進行。一般認為,SR=100%,不存在基體效應;SR 值在80%~120%范圍內(不包括100%),表明有微弱的基質效應;SR 值在50%~80%或120%~150%范圍內,表明有中度基質效應;當SR 值<50%或>150%時,表明有較強的基質效應。
通過對31 個生物堿類成分的分析,發現其中13%對香精油化妝品基質的作用不大;有52%的生物堿具有中度影響;其中16%的生物堿具有中度影響;19%的生物堿類成分有較強的作用。雷公藤次堿、氧化苦參堿、苯甲酰/次/新烏頭堿等具有較強影響。因此,必須采取相應的方法來消除基體的影響。常用的方法有兩種,一種是利用標樣進行校準曲線修正,另一種是利用內標法對其進行校正。
4.4 線性關系、檢出限與定量限
在保障色譜-質譜條件最佳情況下,分別通過不同體積濃度混合標準溶液來測定31 種生物堿的離子對峰面積和質量濃度,并將測定結果分別作為坐標軸的縱坐標、橫坐標,根據形成的坐標軸獲取31 種生物堿的對應回歸方程,相關回歸方程的線性相關性系數均處于0.992 9~1.000 0 區間。根據分析結果可知,31 種生物堿在其對應的線性范圍內均具備較好的線性關系。將混合標準溶液的質量濃度設定為2.5 μg/L,對31 種生物堿的檢出限范圍進行分析計算,獲得對應的檢出限區間為0.11~3.04 μg/kg,31 種生物堿的定量下限區間為0.33~1.13 μg/kg。
4.5 回收率與精密度
以陰性標準溶液為基底溶液,額外添加1 倍、2 倍、3 倍體積濃度的混合標準溶液,充分混合后用于加標回收試驗。通過三種濃度溶液分別開展加標回收試驗,各重復6 次,獲取6 次測試結果后計算每種生物堿的平均回收率均處于63.4%~126.5%,相對標準差處于0.74%~16.81%。總體來說,高效液相色譜-串聯質譜法的回收率與精密度均可以滿足日常檢測標準要求。具體測定結果如表1 所示。
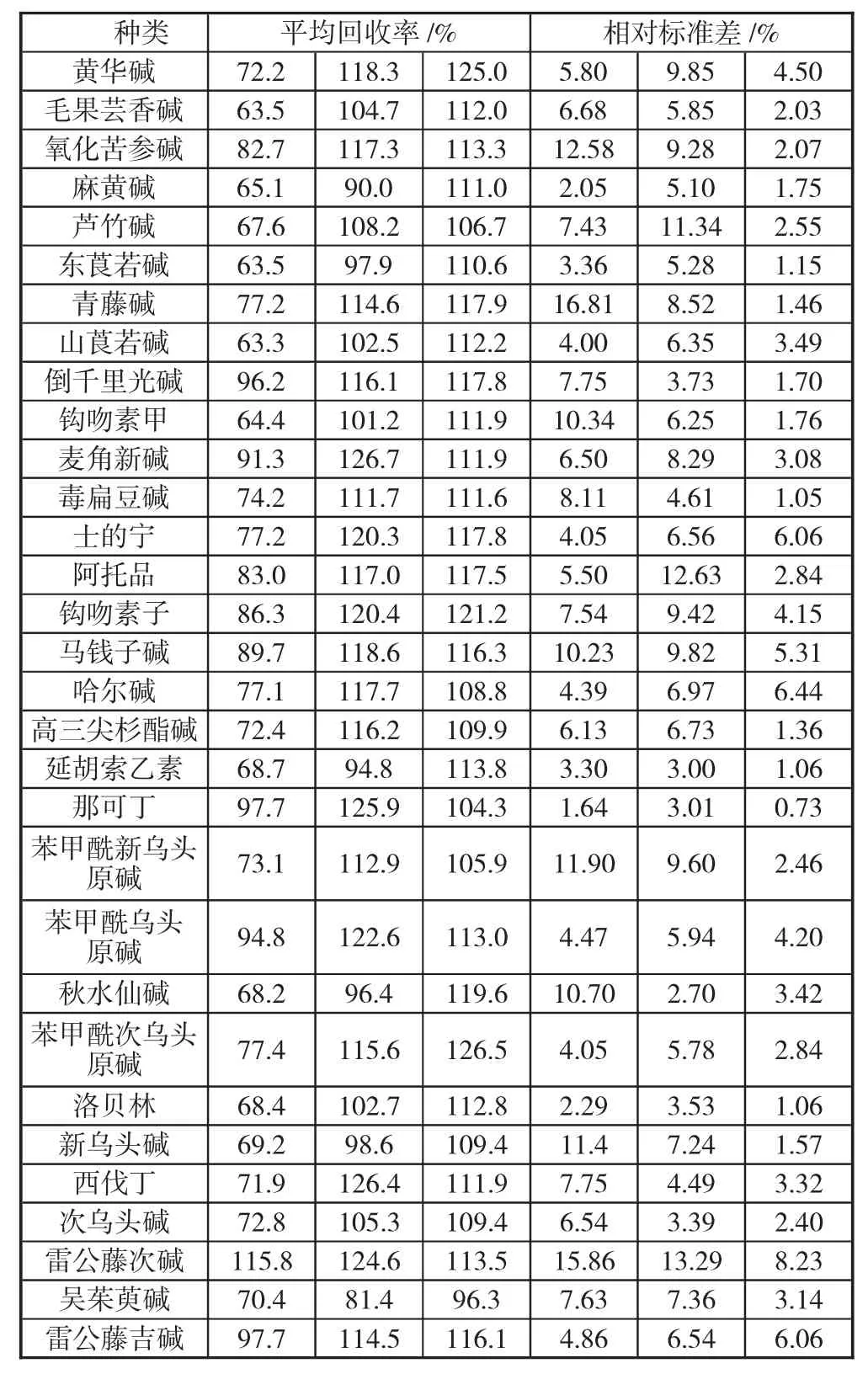
表1 31 種生物堿平均回收率和相對標準差測定結果
4.6 實際樣品的檢測
隨機選取市面上20 種知名精油類化妝品進行檢測,確認所檢驗的20 種化妝品均未檢測中上文中31 種禁用生物堿。同時,實驗中樣品的加標回收率均控制在69.2%~115.6%區間,確認符合質量控制要求。
5 結語
以LC-MS/MS 技術對31 種植物精油中31 種生物堿類成分進行了測定,其中,以含2%甲酸的甲醇-水溶液(體積比為3∶1)為萃取劑,正己烷去油脂,HPH-C18 柱對31 個生物堿成分進行高效分離,經驗證,該方法操作簡便、快速、準確,能滿足31 種植物精油化妝品中禁用生物堿的日常監測需求。
