嬰兒β-酮硫解酶缺乏癥1例
2024-03-22 12:02:00藺艷婷李坤霞陳坤琦張愛梅山東第二醫科大學臨床醫學院兒科教研室濰坊605煙臺毓璜頂醫院兒內科濰坊市人民醫院新生兒科通訊作者mailzamcncom
山西醫科大學學報 2024年2期
藺艷婷,李坤霞,陳坤琦,張愛梅(山東第二醫科大學臨床醫學院兒科教研室,濰坊 605;煙臺毓璜頂醫院兒內科;濰坊市人民醫院新生兒科;通訊作者,E-mail:zamcn@6.com)
β-酮硫解酶缺乏癥(β-ketothiolase deficiency,BKD),又稱線粒體乙酰乙酰基輔酶A硫解酶(T2)缺乏癥,是一種罕見的由乙酰輔酶A酰基轉移酶-1(acetyl coenzyme A acetyltransferase-1,ACAT1)基因變異引起的常染色體隱性遺傳代謝性疾病。ACAT1基因編碼在人類染色體11q22.3-23.1上,橫跨基因組約27 kb的區域,包含12個外顯子和11個內含子[1]。T2缺乏的臨床特征是不同程度的間歇性酮癥酸中毒發作,嚴重者可能出現認知功能損害、神經系統發育落后甚至死亡。現回顧性分析1例經基因診斷確診的BKD嬰兒臨床資料,探索BKD患兒的臨床表現及遺傳特點,進一步提高臨床醫師對該病的認識。
1 病例報告
1.1 病例資料
患兒,女,7月,以“呼吸困難1 d”入院。患兒1 d前無明顯誘因出現呼吸困難,表現為呼吸幅度增大,伴呻吟及輕咳,吃奶少,無發熱,無嘔吐、腹脹、腹瀉,無抽搐及意識障礙。患兒系G2P2,父母體健,無遺傳病病史。有1個哥哥,6歲,體健。
入院體格檢查:呼吸32次/min,心率142次/min,SpO298%,體質量7.5 kg。發育正常,營養欠佳,嗜睡,輕度脫水貌。深大呼吸,雙肺呼吸音粗,未聞及干濕性啰音。心音有力,律齊,各瓣膜聽診區未聞及雜音。腹平軟,肝臟肋下2 cm,脾臟肋下1 cm,腸鳴音正常。
入院輔助檢查:①血氣分析:pH 6.951,PCO210.6 mmHg,PO2130 mmHg,ABE -30.0 mmol/L,SBE -27.7 mmol/L,實際碳酸氫根2.2 mmol/L,標準碳酸氫根5.3 mmol/L,乳酸1.6 mmol/L;②血常規:紅細胞4.21×1012/L,白細胞32.54×109/L,中性粒細胞百分比73.9%,血小板462×109/L;③尿常規:尿蛋白+,尿酮體+++;④總膽固醇5.83 mmol/L(參考值3.12~5.72 mmol/L),甘油三酯2.01 mmol/L(參考值0.40~1.70 mmol/L);⑤甲功:FT4為6.79 pmol/L(參考值12.00~22.00 pmol/L),FT3為1.56 pmol/L(參考值2.8~7.1 pmol/L);⑥胸片:未見異常;⑦顱腦MR:右側基底節區血管間隙擴大。
串聯質譜檢測結果:血串聯質譜遺傳代謝病檢測提示患兒3-羥基異戊酰肉堿(C5-OH)為1.18 μmol/L(參考值0.05~0.70 μmol/L)、異戊烯酰肉堿(C5∶1)為0.24 μmol/L(參考值0.01~0.06 μmol/L)、3-羥基丁酰肉堿(C4-OH)為1.66 μmol/L(參考值0.03~0.75 μmol/L),均顯著增高。尿氣相色譜質譜有機酸檢測提示患兒2-甲基-3-羥基丁酸為22.3 μmol/L(參考值0~4.0 μmol/L)、3-甲基巴豆酰甘氨酸為24.7 μmol/L(參考值0~5.0 μmol/L)、甲基巴豆酰甘氨酸為15.3 μmol/L(參考值0~5.0 μmol/L),均明顯增高。
1.2 基因檢測
本研究經醫院醫學倫理委員會批準(KYLL20230904-4)并獲得患兒監護人知情同意并簽署知情同意書。采集患兒外周血樣3 mL,提取全血基因組DNA,制備文庫,靶向捕獲與富集,利用NovaSeq 6000測序儀進行高通量測序。對候選變異在HGMD、1000 Genomes、dbSNP、gnomAD、ClinVar、ESP 6500等數據庫檢索收錄情況。依據美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)變異評級指南對候選變異致病性進行判讀。結果提示患兒ACAT1(NM_000019.3)基因4號外顯子存在c.253_255del(p.Glu85del)雜合變異,8號外顯子存在c.806_807del(p.Thr269fs)雜合變異(見圖1),均為疑似致病變異。
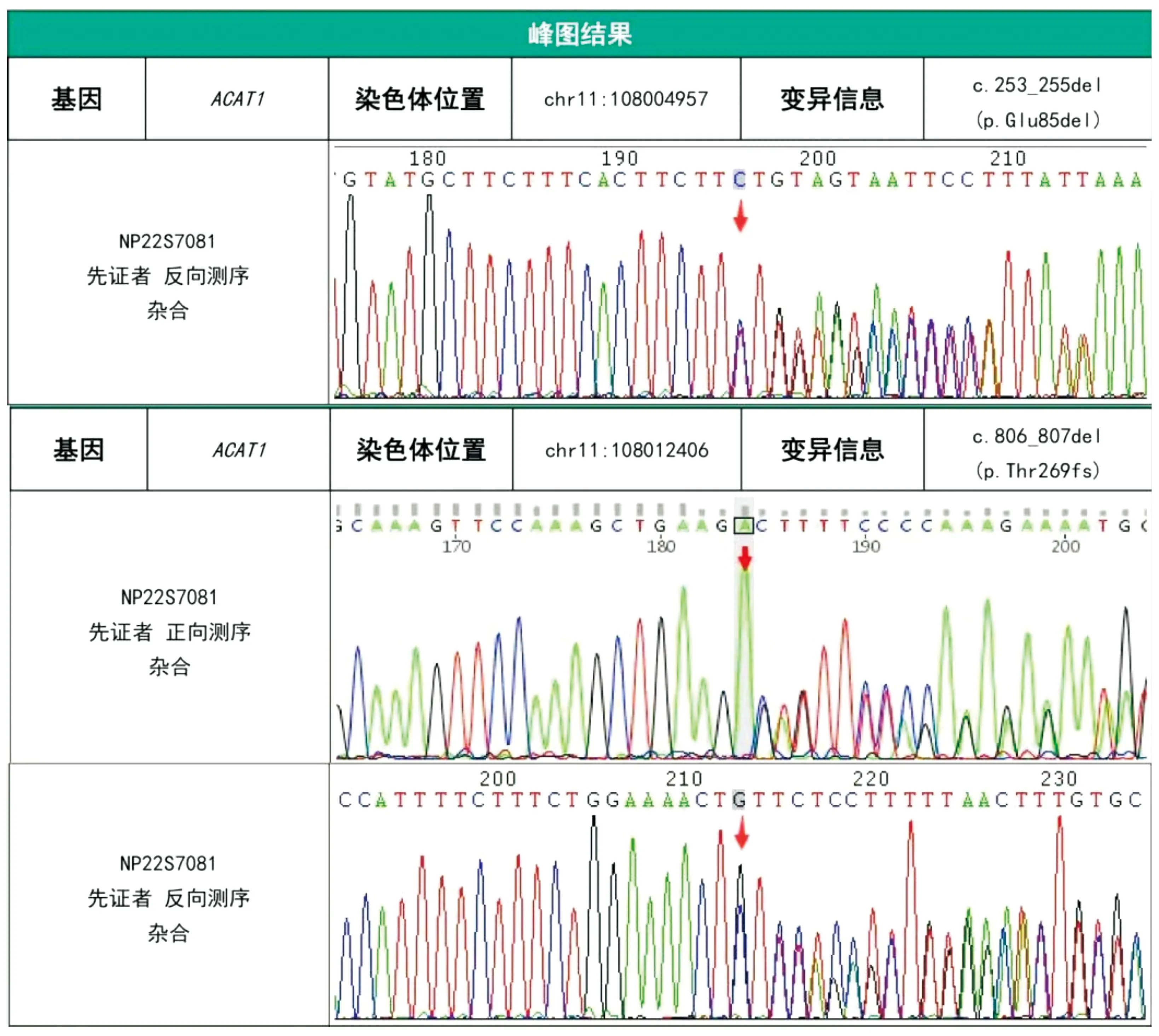
圖1 患兒基因檢測結果
1.3 治療與隨訪結果
該嬰兒入院后給予頭孢哌酮舒巴坦鈉抗感染,碳酸氫鈉糾酸及補液治療。住院期間出現抽搐1次,給予水合氯醛灌腸止痙,急查顱腦CT未見異常。遺傳代謝病檢查結果提示β-酮硫解酶缺乏,給予禁蛋白飲食,無亮氨酸營養粉喂養,加左卡尼汀促進酸性代謝產物排出。患兒住院治療9 d,呼吸困難緩解,復查血氣分析大致正常。出院診斷為:β-酮硫解酶缺乏癥;重度代謝性酸中毒;代謝性腦病;高甘油三酯血癥;急性支氣管炎。出院后予以適當限制蛋白質攝入量,高熱量、低脂飲食,無亮氨酸營養粉喂養,口服左卡尼汀治療,隨訪患兒至2歲無酸中毒發作,生長發育正常。
2 討論
β-酮硫解酶缺乏癥是一種罕見的異亮氨酸分解代謝和酮體代謝異常的先天性疾病。其發病年齡通常為6~18個月。典型表現為由禁食、感染或體力消耗等生酮應激狀態下引起的嘔吐、呼吸困難、呼吸急促、嗜睡或昏迷[2]。在發病前及時干預和治療,可完全恢復或延緩發病時間。BKD的實驗室特征性表現包括顯著的酮尿和尿中異亮氨酸分解代謝中間產物2-甲基-3-羥基丁酸、2-甲基乙酰乙酸和甲基巴豆酰甘氨酸的排泄增加。血串聯質譜檢測通常顯示異戊烯酰肉堿和3-羥基異戊酰肉堿水平升高。有研究表明3-羥基丁酰肉堿同樣是檢測BKD的一個非常強大的標記物[3]。
本例患兒因呼吸困難入院,血氣分析示重度代謝性酸中毒合并失代償性呼吸性堿中毒,尿常規示酮體+++,住院期間出現抽搐,血串聯質譜示C5-OH、C5∶1、C4-OH顯著增高,尿氣相色譜質譜示2-甲基-3-羥基丁酸、3-甲基巴豆酰甘氨酸、甲基巴豆酰甘氨酸明顯增高,確診為β-酮硫解酶缺乏癥。基因檢測示患兒ACAT1基因4號外顯子c.253_255del為框內缺失變異,導致所編碼蛋白質第85位氨基酸Glu發生缺失,但不會引起移碼。有文獻報道在乙酰乙酰基輔酶A硫解酶缺乏癥患者中檢測到該變異[1],該變異在大規模人群頻率數據庫gnomAD中有2例雜合子報道,未見純合子報道。功能學研究表明,該變異導致線粒體乙酰乙酰基輔酶A硫解酶活性顯著降低。依據ACMG指南評級為疑似致病變異(PM2+PM4+PS3_Moderate+PM3_Supporting)。ACAT1基因8號外顯子c.806_807del是非3倍數堿基缺失導致的移碼變異,理論上可通過無義介導的mRNA降解(NMD)或編碼氨基酸序列的提前終止,而導致正常蛋白功能喪失。該變異在HGMD、dbSNP、ESP 6500、1000 Genomes等數據庫均未見收錄。依據ACMG指南評級為疑似致病變異(PVS1+PM2)。患兒父母拒絕對基因變異進行溯源,因此不能明確突變來源。患兒父母臨床表型均未見異常,考慮為隱性攜帶者的可能性大。我們推測患兒的框內缺失變異及移碼變異分別來自父母,結合患兒臨床表型、家族史、串聯質譜檢測及基因檢測結果,考慮患兒為ACAT1基因復合雜合突變所致的β-酮硫解酶缺乏癥。
BKD的發病率各個國家報道不一,無明顯的種族特異性,越南北部的發病率估計為1∶190 000[4],北卡羅來納州為1∶313 000[3],美國明尼蘇達州為1∶232 000[5]。中國的報道多為散發病例,少見流行病學的相關分析資料。在浙江省新生兒疾病篩查中心的一項多中心國家隊列研究估計中國的發病率最低約為1∶1 000 000。迄今為止,至少有105種與BKD相關的ACAT1基因變異報道[6]。最常見的變異類型是錯義突變。基因變異的類型在不同國家和種族之間有所不同。越南北部的熱點變異為c.622C>T(p.Arg208X)[7],印度的熱點變異為c.578T>G(p.Met193Arg)[4]。徐烽等[8]報道c.1124A>G(p.Asn375Ser)變異可能是中國人的熱點變異。值得一提的是,本例患兒c.806_807del變異在中國知網及PubMed等數據庫均未見相關報道,為新發變異。
ACAT1基因編碼的線粒體乙酰乙酰輔酶A硫解酶(T2)在異亮氨酸分解中使2-甲基-乙酰乙酰輔酶A分解為乙酰輔酶A和丙酰輔酶A,T2在酮體利用過程中催化乙酰乙酰輔酶A生成2分子乙酰輔酶A[9]。BKD患兒T2活性降低或喪失,異亮氨酸分解代謝障礙,導致大量酸性代謝產物蓄積,同時大量酮體在組織細胞中聚集。因此患兒常反復發生嚴重代謝性酸中毒及多臟器損害。本例患兒血氣分析示重度代謝性酸中毒,因診斷及治療及時,尚未發現有嚴重的臟器損害。因此,早篩查、早診斷及合理治療是挽救患兒、改善預后的關鍵。我們要積極普及新生兒遺傳代謝性疾病篩查,以期在急性代謝性酸中毒首次發作之前得到及時、恰當的診治。我們應該警惕出現嚴重代謝性酸中毒、不明原因的昏迷或神經系統發育落后的患兒發生遺傳代謝性疾病的可能,及時送血尿標本行串聯質譜檢測,以盡早明確診斷。
目前對于無癥狀或非發作性癥狀的BKD患兒,應避免禁食,建議口服L-肉堿,補充劑量為50~200 mg/(kg·d)。若患兒處于生酮狀態,如嘔吐、食欲減退或感染等,即使血糖正常,也應口服或靜滴補充充足的葡萄糖,以抑制酮體的生成。在密切監測血糖的同時,使用胰島素(1 U)與足量的葡萄糖(4~6 g)配制靜脈輸注,可預防患兒發生嚴重的酮癥酸中毒[10]。同時根據個體情況補充碳酸氫鈉。嚴重患兒需要血液透析或血漿置換。處于穩定期的患兒需限制蛋白質。有文獻報道[11],建議嬰兒蛋白質攝入量控制在1.5~2.0 g/(kg·d),1歲以上的患兒蛋白質攝入量控制在1.0~1.5 g/(kg·d)。同時注意避免高脂飲食[12]。口服左卡尼汀有助于穩定代謝狀況,左卡尼汀可與有機酸代謝產物形成酰基肉堿,有利于有機酸的排泄。
關于基因型與表型的關系,以往的研究表明,基因型與臨床表型在BKD中并不相關。即使BKD患者具有相同的基因型和相似的環境因素,也可能具有不同的臨床表型。雖然基因型可能影響BKD患者的生化表型,但由于缺乏對大量家族性ACAT1變異的研究,基因型與生化表型之間的關系難以分析[3]。
綜上所述,本研究通過基因檢測對1例BKD嬰兒進行了精準的分子學診斷,確診其為ACAT1基因c.253_255del及c.806_807del復合雜合變異所致的β-酮硫解酶缺乏,為該嬰兒的后續治療及家系遺傳咨詢提供了理論依據。新變異的檢出同時拓展了ACAT1基因的變異譜。
